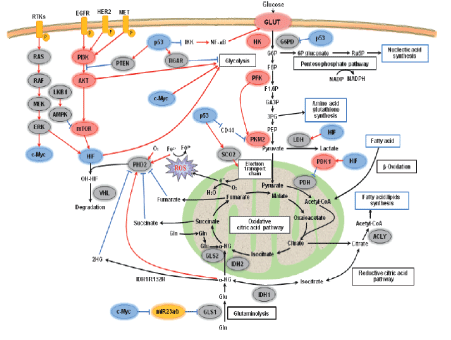
Figure 1: Signaling networks and their regulation of metabolism in cancer cells.
Abraham Nigussie Mekuria*
School of Pharmacy, College of Health and Medical Sciences, Haramaya University, Ethiopia
*Corresponding author: Abraham Nigussie Mekuria, School of Pharmacy, College of Health and Medical Sciences, Haramaya University, Ethiopia, Tel: 251911194519; E-mail: abrishn@yahoo.com
Transformed cells undergo a metabolic transformation to satisfy the demands of growth and proliferation. In this regard, cancer cells prefer to perform glycolysis in the cytosol even in the presence of oxygen, a phenomenon first observed by Otto Warburg and now famously known as “Warburg effect” or “aerobic glycolysis”. Such reprogramming of glucose metabolism has been validated within many tumors, and increased glycolysis facilitates biosynthesis of biomass (e.g., nucleotides, amino acids and lipids) by providing glycolytic intermediates as raw material. Besides the dysregulation of glucose metabolism, metabolic reprogramming in cancer cells has been characterized by aberrant lipid metabolism, amino acids metabolism, mitochondrial biogenesis, and other bioenergetics metabolic pathways. However, the two noticeable characteristics of tumor cell metabolism are the Warburg effect and glutaminolysis, which, respectively, demonstrate the dependence of tumor cells on glucose and glutamine. This review aimed at appraising recent findings related to the drivers of glucose and glutamine metabolism reprogramming, their crosstalk in cancer cells, and their potential in cancer therapy.
Metabolic reprogramming; Novel targets; Cancer; Glycolysis; Glutaminolysis
Compared to normal cells, tumor cells show an essentially contrarily accustomed metabolism so as to find ways to proliferate, even though both types of cells use the same nutrients [1,2]. Otto Warburg showed that cancer cells are addicted to glycolysis; they ferment glucose into lactate rather than committing into mitochondrial oxidative phosphorylation (OXPHO), regardless of oxygen tension [3]. He postulated that defect in tumor cells mitochondria resulted in reduced OXPHO [4]. But, this is not the case, according to current understanding. They do, however, adapt their function to the needs of cell proliferation. Mitochondria, in addition to acting as a hub for ATP production, it serves a significant role by synthesizing precursors required for proteins, lipids, and nucleic acids synthesis via Krebs’s cycle [5-7]. To this end, in the following sections recent findings related to the drivers of glucose and glutamine metabolism reprogramming, their crosstalk in cancer cells, and their potential as cancer therapeutic strategy will be reviewed.
According to, existing literatures pointed out that alterations in numerous signaling pathways and altered expression and mutation of metabolic enzymes are central in mediating the unusual metabolic behavior of cancer cells [8-10].
Proliferating cells, i.e. both cancer cells and normal cells exhibit metabolic reprogramming, but, in normal cells, growth factor (GF) signaling-induced alterations to metabolism are responsive to environmental signals and rapidly down-regulated if circumstances are unfavorable for growth [8]. In contrast, in tumor cells, internal and external cues turn out to be decoupled, owing to up-regulation of oncogenic signaling pathways and/or down regulation of tumor suppressor signaling pathways [9] (Figure 1).
It shows various aspects of energy metabolism regulation, including glycolysis, TCA cycle, pentose phosphate, glutaminolysis, fatty acid biosynthesis pathway, PI3K and RAS-MAPK signaling cascade. Three transcription factors, HIF-1, c-Myc and p53, are key regulators and coordinate regulation of cancer metabolism in different ways. 2HG, 2-hydroxyglutarate; 3PG, 3-phospho-glycerate; 6P gluconate, 6-phospho-gluconate; a-KG, a-ketoglutarate; ACLY, acetyl-CoA by ATP-citrate lyase; AKT, v-akt murine thymoma viral oncogene homologue; AMPK, AMP-activated protein kinase; CD44, is a glycoprotein; EGFR, epidermal growth factor receptor; F1,6P, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; G6P, glucose-6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; Gln, glutamine; GLS, glutaminase; Glu, glutamic acid; GLUT, glucose transporter; HER2, human epidermal growth factor receptor type 2; HIF, hypoxia-inducible factor; HK, hexokinase; IDH, isocitrate dehydrogenase; IKK, nuclear factor-j light-chain-enhancer of activated B cells kinase; LDH, lactate dehydrogenase; LKB1, liver kinase B1; MET, hepatocyte growth factor receptor; mTOR, mammalian target of rapamycin; NF-jB, nuclear factor-j light-chainenhancer of activated B cells kinase; OH, hydroxy; P, phosphate; PDH, pyruvate dehydrogenase; PEP, phosphoenolpyruvate; PFK, phosphofructokinase; PHD2, prolyl hydroxylase 2; PI3K, phosphatidylinositol 3-kinase; PKM2, pyruvate kinase isozyme type 2; PTEN, phosphate and tensin homolog deleted on chromosome 10; RAF, regulation of alpha-fetoprotein; RAS, rat sarcoma virus peptide; ROS, reactive oxygen species; RTK, receptor tyrosine kinases; Ru5P, ribulose-5-phosphate; sCO2, synthesis of cytochrome c oxidase 2; TCA, tricarboxylic acid; TIGAR, Tp53-induced glycolysis and apoptosis regulator; VHL, von Hippel-Lindau tumor suppressor, adapted from Song [9].
The Phosphoinositide-3-Kinase (PI3K): Pathway: PI3K is one of the most commonly rearranged signaling pathways in human cancer cells [11]. It could be because of mutation in phosphate and tensin homolog (PTEN), a tumor suppressor gene that inhibits the PI3K pathway [12]. Besides, mutations in the components of the pathway itself have also been associated with PI3K activation [13]. Abnormal signaling through receptor tyrosine kinases (RTK) upstream to the PI3K pathway have been also associated with aberrant activation of the PI3K pathway [12]. In this regards, activation of the pathway directly affects cellular metabolism like stimulation of glycolysis possibly via up regulating glucose transporters proteins expression and membrane translocation, as well as by activating crucial glycolytic enzymes via phosphorylation as shown in figure 1 [9,14]. Besides, indirectly through activation of mammalian target of rapamycin (mTOR) [15], that is known to regulate transcription factors such as hypoxiainducible factor-1 (HIF1) leads to HIF1-dependent metabolic changes as shown in figure 1 and table 1 [9,16,17].
| Pathways | Target genes | Transcription factors |
| Transporter | Glucose transporter 1 | HIF, c-Myc & p53 |
| Glucose transporter 2 | c-Myc | |
| Glucose transporter 3 | HIF & p53 | |
| Glucose transporter 4 | c-Myc & p53 | |
| Glycolysis | Hexokinase 2 | HIF, c-Myc & p53 |
| Phosphofructokinase 1 | HIF & c-Myc | |
| Aldolase A | HIF & c-Myc | |
| GAPDH | HIF & c-Myc | |
| Phosphoglycerate kinase 1 | HIF & c-Myc | |
| Phosphoglycerate mutase | p53 | |
| Enolase 1 | HIF & c-Myc | |
| Pyruvate kinase M2 | HIF & Myc | |
| Lactate dehydrogenase A | HIF & c-Myc | |
| Pentose phosphate | Transketolase | HIF |
| Transketolase-like protein 2 | HIF | |
| TCA cycle | Pyruvate dehydrogenase kinase 1 |
HIF & c-Myc |
| Glutaminase 2 | p53 | |
| Others | Carbomyl phosphate synthetase aspartate transcarbomylase & dihydroorotase | c-Myc |
| Serine hydroxymethyl transferase |
c-Myc | |
| Fatty acid synthase | c-Myc | |
| Ornithine decarboxylase | c-Myc |
Table 1: Target genes of HIF, c-Myc and p53 associated with energy metabolism [9].
Liver Kinase B1 (LKB1)/Adenosine Monophosphate-Activated Protein Kinase (AMPK) Pathways: Activation of the pathway known to regulate energy metabolism and growth, stimulating gene expression for extensive changes in metabolic programming, suppressing protein synthesis, and stimulating fatty acid oxidation to replenish ATP [18,19]. For instance, AMPK directly phosphorylates peroxisome proliferator activated receptor gamma (PPAR-γ) coactivator-1-α (PGC-1α), a transcriptional coactivator that controls several metabolic genes and mitochondria formation (Figure 1) [20]. However, loss of activity of AMPK has been associated with promotion of carcinogenesis via increasing the glycolytic pathway in tumor cells. This promotes a metabolic shift toward the Warburg effect [21]. Moreover, loss of LKB1 expression in tumor cells reduces the AMPK signaling, making cells more sensitive to low nutrient level, and leading to unregulated metabolism and cell growth in energetically stressful conditions [22-25]. This might promote tumorigenesis, as it leads to elevated glucose and glutamine flow, rising ATP levels, and a metabolic switch to aerobic glycolysis.
Figure 1: Signaling networks and their regulation of metabolism in cancer cells.
Hypoxia-Inducible Factor-1: HIF1 has been recognized as a key mediator of metabolic response to hypoxia [9]. It is a heterodimer composed of constitutive, stable β subunits and unstable α subunits, which are synthesized yet, degraded under presence of adequate oxygen due to the sequential action of oxygen-dependent prolyl hydroxylases (PHDs) and the VHL ubiquitin ligase (Figure 1). It functions as a transcriptional activator and enhances expression many oncogenes, including vascular endothelial growth factor (VEGF), which promotes angiogenesis; epidermal growth factor (EGF); insulin like growth factor-2 (IGF-2); transforming growth factor beta (TGF-β) [26], which stimulates growth and cell survival, and most importantly reprogram energy metabolism as shown in table 1 [9].
Myc: There is difference between normal cells and cancer cells in the level of expression of Myc [10]. According to studies, expression of Myc is induced by GF stimulation in normal cells, whereas in cancer cells there is over expression of Myc without regarding to GF signaling and this over expression is estimated to occur in 70% of human tumors [27,28]. Subsequently, over activity of Myc stimulates energy generation and precursor synthesis required for fast proliferation tumor cells [10]. Similar to HIF, Myc reprogram energy metabolism by altering target gene expression (Table 1).
p53: It plays an essential part in regulating the activities of glycolysis and OXPHOS (Table 1), in addition to its role in DNA damage response and apoptosis [29]. In general, p53 decreases the glycolytic rate, however, mutation or suppression of p53 frequently occurs in cancer, which results in losing control of its functions, thus promoting glycolysis. Surprisingly, mutant p53 inhibit mitochondrial respiration by down-regulating expression of cytochrome c oxidase 2 (sCO2 ) and Glutaminase 2 (GLS2) [30]. Moreover, it activates AKT and HIF, which are effectors downstream of PI3K [31].
Bcl-2 Proteins: Accumulated body of evidence has shown the involvement of the apoptotic mediator, B cell lymphoma/leukemia-2 (Bcl-2) proteins in reprogramming cancer cells metabolism [32- 34]. A study done by Danial et al. [32] reported integration between glycolysis and apoptosis pathway due observation of mitochondria associated glucokinase (in the liver) with the pro-apoptotic protein Bcl-2/Bcl-xL-associated agonist of cell death (BAD). The study revealed that, glucokinase activation via direct interaction with BAD especially in response to phosphorylation of BAD by Akt, downstream of PI3K pathway. However, glucokinase inhibits BAD’s pro-apoptotic activity when it bounds with BAD in its phosphorylated form. But, dephosphorylated BAD will dissociate from it, and able to interact with the anti-apoptotic protein Bcl-2/Bcl2-like 1, L isoform (Bcl-xL) and stimulate programed cell death. In this regard, binding of BAD to mitochondria associated glucokinase stimulate glucokinase and glycolysis activity that could be considered as one driver of metabolic reprograming in cancer cells, in addition to preventing its proapoptotic functions [33-35].
Furthermore, a pro-apoptotic BH3-containing protein known as damage protein (NOXA) also play a part in metabolic control. According to a study done by Lowman et al. [36] , when there is elevation in of glucose level, NOXA will be phosphorylated by cyclin dependent kinase 5 (CDK5) that leads to localization of this pro-apoptotic protein within the cytoplasm and making it unable to accomplishing its pro-apoptotic functions. As the study found out the protein rather form complex with the anti-apoptotic Bcl-2 protein myeloid cell leukemia-1 (Mcl-1) and stimulates improved glucose metabolism and enhances metabolism via the PPP, favoring synthesis ribose sugar and NADPH. Furthermore, subsequent studies showed that over expression of NOXA in tumor cells, and over activity of CDK5 to promote tumor growth and survival, specifically in thyroid and neuroendocrine tumors [37,38].
In addition to activation of oncogenes and loss of tumor suppressor pathways, mutations in key metabolic enzymes as well as preferential expression of specific isoforms of metabolic enzymes can provide cancer cells a mechanism to select for metabolic alterations during tumorigenesis [1,2,39].
Pyruvate Kinase M2: Recent studies reported that, PK plays a crucial role in reprogramming of glycolytic metabolism. Four mammalian PK isoenzymes (M1, M2, liver isoform (L) and RBC isoform (R)) have been identified and distributed in diverse cell types [40]. The muscle isoform (PKM1) is a constitutively active tetrameric form that is found in normal adult cells, whereas PKM2 forms less active dimers as well as tetramers and found in differentiated tissues and normal proliferating cells [10].
To form the active tetramer, PKM2 requires fructose-1, 6- bisphosphate (F-1, 6 BP). Its tetramer form has high affinity to PEP and leads to improved production of pyruvate [41]. Meanwhile, studies done using cancer cells pointed out that, PKM2 conversion from the tetramer to less active dimer by phosphorylation mediated tyrosine kinases by at tyrosine 105 sites in the enzyme that leads to a conformational change and dissociation of F-1, 6 BP. The PKM2 conformational change caused by phosphorylation leads to FBP release and conversion of the enzyme from the tetramer to the less active dimer form [42,43]. Hence, in tumor cells, PKM2 is predominantly available in its less active dimeric form, this leads to accumulation of glycolytic intermediates upstream to PK. Subsequently, it causes diversion of these intermediates into anabolic pathways which hasten active proliferation of cancer cells as shown in figure 1 [9,41]. In contrast, replacement of embryonic and tumor isoform (PKM2) by PKM1 in tumor cell lines renders them less glycolytically active and diminishes tumor xenograft growth, suggesting that PKM2 might be responsible for the Warburg effect [43,44].
On the other hand, PKM2 has been shown to support tumor growth via “non-metabolic” attributes [45-47]. For instance, in a study done by Luo et al. [45], PKM2 shown to interact with HIF1α within the nucleus and as reported by the study this interaction enhances transcriptional activity of HIF1α. This in turn leads to enhanced expression of target genes, including, GLUT1, PKM2, and LDHA. It is therefore, the study revealed a “positive feedback loop” mechanism that reprograms the glucose metabolism. Similarly, Yang et al. [46] showed that, activation of EGFR resulted in translocation of PKM2 into nucleus where it is associated with phosphorylated β-catenin to form a complex, which enhanced cyclin D1 and c-Myc expression. These findings underscore the importance of the integrated metabolic and non-metabolic functions of PKM2 in tumorigenesis.
Isocitrate Dehydrogenase (IDH): IDH mutations can be seen as a case where a single point mutation (R132) affecting cellular metabolism is selected in cancer cells. In fact, IDH1 mutations were recognized in gliomas and acute myeloid leukemias (AML) [47,48]. It has been known that oxidative decarboxylation of isocitrate by nonmutant IDH1 generates α-ketoglutarate (α-KG) and NADPH, but not the case concerning the mutant IDH1 [49]. In this regard, Dang et al. [50] using in human malignant gliomas revealed that, the mutant IDH1 reduces α-KG to 2-hydroxyglutarate (2-HG) by consuming NADPH rather than generation. In AML, both the cytosolic IDH1 and the mitochondrial analogue IDH2 are commonly mutated [51]. One of the consequences of this change regarding tumorigenesis is that, stabilization of the oncogene HIF-1α, since for its degradation α-KG is required by PDH2 [26]. Moreover, 2-HG was shown to act as a competitive inhibitor of α-KG-dependent demethylases, including histone demethylases and the TET family of 5-methylcytosine hydroxylases, affecting CpG island hypermethylation. This links the oncogenic effect of IDH1 mutations to epigenetic regulation [52,53].
Succinate Dehydrogenase and Fumarate Hydratase: It has been known that, Krebs’s cycle enzymes SDH and FH catalyze the conversion of succinate to fumarate and fumarate to malate, respectively. But, mutant form of these enzymes has been associated with carcinogenesis [54]. In this regard, Pollard et al. [55] reported frequent germline mutation in FH regarding familial cancer syndromes, renal, skin, and uterine cancers. In the same study, mutations in these enzymes caused accumulation of their substrate and these substrates i.e. fumarate and succinate ones accumulated can act as oncogenes when the traverse the inner mitochondrial membrane and enter the cytosol by dioxygenases and prolyl hydroxylases, which are known to be involved in the degradation of the oncogene HIF-1α under normoxic environment [2].
During the past decade, the metabolic rewiring of cancer cells has been viewed as a promising source of novel drug targets (Table 2).
| Metabolic enzyme or transporter protein | Alteration in cancer cells |
Consequence of alteration | Possible drivers | Example cancer types | Compounds under investigation |
References | |
| Glucose transporters | Overexpression of GLUT-1, -3, -4 & -12 |
Facilitate glucose uptake by cancer cells | Over activity of MYC,AKT, HIF-1α, & LOF mutation of p53 |
Brain, breast, head, neck, bladder, renal, colorectal, lung, gastric, ovarian, OED, OSCC, & laryngeal | Phloretin, WZB117, Fasentin |
[56-60] | |
| Hexokinase | Over expression of HK II | Facilitate glucose metabolism & also functions as a protective signaling molecule | Over activity of MYC, & AKT | Breast, colon, lung, liver, ovarian, cervical, pancreatic, glioblastoma, & thyroid |
2-DG | [61-64] | |
| Under expression of HK I | Accelerate tumor growth & metastasis |
Over activity of MYC, & AKT | Cervical | [61] | |||
| Phosphofructokinase 1 | Over expression of pfkfb-3 | Increased production of F2, 6BP, a potent allosteric activator of PFK-1 | Over activity of MYC, & AKT | Breast, colon, ovarian, thyroid, head, neck & squamous cell | PFK158 | [67,68] | |
| Pyruvate kinase | Over expression of PKM2 | Causes accumulation & diversion of glycolytic intermediates upstream to PK into anabolic pathways; enhances transcriptional activity of HIF1α |
Over activity of HIF, EGFR & LOF mutation of p53 |
Lung, liver, colon, thyroid, kidney & bladder |
TLN-232/CAP- 232, Lapachol |
[41-46,69] | |
| Pyruvate dehydrogenase kinase | Over expression of PDK1-3 |
Reduce flux of pyruvate into mitochondria |
Over activity of MYC, HIF-1α, & LOF mutation of p53 |
glioblastoma, breast, melanoma, cervical, colon, & ovarian, |
DCA | [73-77] | |
| Lactate dehydrogenase | Over expression of LDH-A | Prevent buildup of lactate inside cancer cell | Over activity of MYC, HIF-1α, & LOF mutation of p53 |
Liver, colon, lung, & pancreatic | FX11 | [34,70,71] | |
| Monocarboxylate transporters |
Over expression of MCT1 & MCT4 | Facilitate lactic acid effuse from tumor cells | Over activity of MYC & LOF mutation of p53 |
Prostate, gastric, lung, breast, colon | α-cyano- 4-hydroxy- cinnamic acid |
[70-72] | |
| Glutamine transporter proteins | Over expression of SLC1A5 & LAT1 | Sustain glutamine need of cancer cells | Over activity of MYC & LOF mutation of p53 |
Breast, colon, lung , melanoma, neuroblastoma, glioblastoma, & prostate |
KM8094, BCH, GPNA | [83-88] | |
| Glutaminase |
Over expression of GLS1 |
Maintain a functioning TCA cycle | Over activity of MYC, KRAS, Rho GTPases & LOF mutation of p53 |
Colon, breast, lung, cervix, brain; human B lymphoma, prostate, acute myeloid leukemia, myeloma, & gliomas | BPTES, CB- 83958, & compound 968 |
[89-96] | |
| Glutamate dehydrogenase |
Over expression of GLUD | Maintain a functioning TCA cycle | Over activity of MYC |
Gliomas, leukemias, breast, lung & colon | EGCG, R162 | [74,96] | |
| Isocitrate Dehydrogenase |
GOF mutation of IDH1, & IDH2 | Production of 2HG from α-KG & resulted in stabilization of HIF-1α |
- | Gliomas & acute myeloid leukemias |
AG-221 | [48-51] | |
| Succinate Dehydrogenase & Fumarate Hydratase | LOF mutations FH, SDH B, -C & -D | Increased succinate &/ or fumarate causes stabilization of HIF-1α | - | Renal, skin, & uterine | - | [54,55] | |
Table 2: Altered enzymes and transporter proteins in glucose and glutamine metabolism and possible drivers in various types of cancers.
Glucose transporter (GLUT); fructose-2, 6,-bisphosphate (F2, 6BP); pyruvate kinase (PK); 2-deoxyglucose (2-DG); Dichloroacetate (DCA); Hexokinase (HK); Loss of function (LOF); Gain-of-function (GOF); oral epithelial dysplasia (OED); oral squamous cell carcinoma (OSCC); Pyruvate dehydrogenase kinase (PDK).
2-hydroxyglutarate (2HG); α ketoglutarate (α-KG); Succinate Dehydrogenase (SDH); Fumarate Hydratase (FH); Isocitrate Dehydrogenase (IDH); Solute carrier family A1 member 5 (SLC1A5); L-type amino acid transporter 1 (LAT1); Glutaminase 1 (GLS1); Monocarboxylate transporters (MCT); Lactate dehydrogenase (LDH); 2-aminobicyclo-(2, 2,1)-heptane-2-carboxylic acid (BCH); gamma-l-glutamyl-p-nitroanilide (GPNA); bis-2-[5–phenylacetamido-1, 2, 4-thiadiazol-2-yl] ethyl sulfide (BPTES), Epigallocatechin gallate (EGCG); Glutamate dehydrogenase (GLUD); Loss of function (LOF); Gain-of-function (GOF).
Targeting Glucose Metabolism: As shown in table 2, targeting GLUTs, HK-II, PFK-1, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), PKM2, and Krebs’s cycle mutant enzymes has been tried as part of development of anticancer drugs to modulate glucose metabolism in tumors [56-79]. For instance, several compounds, including, phloretin, WZB117 and fasentin has been demonstrated antitumor effects in preclinical studies by inhibiting GLUTs. However, selectivity of such drugs against tumors is under question because they are ubiquitously expressed in mammalian cells [56]. Moreover, 2-deoxyglucose (2-DG), a glucose analogue has been identified as a small molecule that inhibits HK and glycolysis according to in vitro and in vivo studies as reviewed by Xi et al. [65]. Furthermore, in study done by Zhu et al. [66] 2-DG showed in improved inhibition of growth, migration, invasion and cell cycle arrest when combined with metformin against ovarian cancer cell lines via p38 MAPK/JNK signaling pathway.
On the other hand, mutation in the Krebs’s cycle enzymes IDH, FH, and SDH have been identified in different cancer types [78,74]. Novel compounds like that target the gain-of-function activity of mutant IDH have recently been shown to have success in preclinical and clinical settings [78], however, inhibiting mutant FH and SDH with small molecules has been unrealistic because these are loss of function mutations [74]. Accordingly, AG-221, inhibitor of mutant IDH2 has been shown to decrease the production of 2HG and cause tumor cells to differentiate towards a more normal phenotype and it is early phase clinical trials [79].
Targeting Glutamine Metabolism: The idea of interrupting the supply or utilization of the conditionally-essential amino acid glutamine in order to fight cancer dates back several decades and is based on its high concentration in plasma as well as the selective vulnerability of a variety of malignant cells to glutamine depletion [80,81]. In this regard, studies has been investigating several small molecules which inhibits glutamine transporter proteins and glutaminase enzyme that play a great role in cancer cell glutamine metabolism as shown in table 2 [82-94].
For instance, it has been recognized that, solute carrier family A1 member 5 (SLC1A5) and L-type amino acid transporter 1 (LAT1) which are involved glutamine transport in the cell shown to be upregulated in malignancies [82,83]. To inhibit glutamine uptake by tumor cells different compounds have been tested in vitro and in vivo [84]. In a study done by Hassanein et al. [85] aimed at evaluating SLC1A5 as a potential target and candidate biomarker predictive of survival and response to therapy, targeting was examined in a panel of NSCLC and human bronchial cell lines by RNA interference and by a small molecular inhibitor, gamma-l-glutamyl-p-nitroanilide (GPNA). In the study, inactivation of SLC1A5 genetically or pharmacologically has been shown to decrease glutamine consumption, inhibit cell growth, and also induce autophagy and apoptosis in a subgroup of NSCLC cell lines that over express SLC1A5. Moreover in the same study targeting SLC1A5 has been shown to decrease tumor growth in NSCLC xenografts. Similarly, in a recent study reported by Kasai et al. [86] has been the anti-tumor efficacy of a novel anti-SLC1A5 humanized monoclonal antibody, KM8094 against gastric cancer by inhibiting glutamine uptake. On the other hand, a study done by Imai et al. [87] using inhibitor of LAT1, 2-aminobicyclo-(2, 2,1)-heptane2-carboxylic acid (BCH), demonstrated reduction in viability of on-small cell lung cancer cell lines as well as, co-administration of gefitinib with BCH reduced the viability of the cells more than either agent alone. The authors reported that inhibition of LAT1 reduced the level of phosphorylation of mTOR, p70S6K and 4EBP1.
Moreover, it is known that GLS is required to generate glutamate from glutamine during glutamine metabolism is GLS [1]. GLS has been inhibited using small molecule inhibitors such as bis-2-[5– phenylacetamido-1, 2, 4-thiadiazol-2-yl] ethyl sulfide (BPTES), CB83958 and compound 968 [88-90]. In these studies inhibition has been shown to significantly suppress tumor growth in several experimental models including breast cancer and lymphoma. Moreover, a recent study done by Song et al. [91] demonstrated that, loss of GLS1 expression by RNAi shown to decrease proliferation and survival of colorectal cancer (CRC) cells due to decrease in ATP levels and increases ROS level.
However, in a study done by Cheng et al. [89] silencing of GLS inhibits cell proliferation but fails to eliminate glioblastoma cells in both in vitro and in vivo models. The same study found out that induction of a compensatory anaplerotic mechanism mediated by pyruvate carboxylase (PC), allows the tumors to use glucosederived pyruvate instead of glutamine for anaplerosis. Furthermore, Phannasil et al. [92] reported that expression of PC in cancerous areas of breast tissue at higher levels than in the non-cancerous areas by examining the expression of PC using Immunohistochemistry of paraffin-embedded breast tissue sections of fifty seven breast cancer patients with different stages of cancer progression. In this regard, dual targeting of both GLS and PC could produce synergistic activity in arresting growth of tumors having glutamine addiction.
Realizing the intricate nature of metabolic links and how different tumors adjust these processes to satisfy their metabolic demands will be one of the most important challenges in exploiting cancer metabolism target for cancer therapy. In this regard, explicit knowledge regarding most feasible targets and there control and cross-talk at different levels of regulation will transform the efforts of current studies in to fruit i.e. producing a successful anticancer agent targeting cancer metabolism. The other issue that could be a challenge and should be addressed in the future is selectivity, because highly proliferating cells like T lymphocyte cells have similarity in metabolic profiles like cancer cells, it is therefore, understanding the critical difference between cancer and highly proliferating normal cells will have paramount importance in avoiding toxicity. On the other hand, combining metabolic inhibitors with the currently available drugs which have been associated with cell death via oxidative stress, might leads to synergistic effect by arresting pro-survival mechanisms via generation of ATP as well as reducing powers like NADPH via PPP.
The author declares that there is no conflict of interests regarding the publication of this paper.
The author would like to thank Professor Ephrem Engidawork for his help and making this review possible.
Download Provisional pdf here
Article Type: REVIEW ARTICLE
Citation: Mekuria AN (2018) Driver of Glucose and Glutamine Metabolism Reprogramming in Tumor Cells and Their Potential as a Target for Cancer Therapy. Int J Cancer Res Mol Mech 4(1): dx.doi.org/10.16966/2381-3318.141
Copyright: © 2018 Mekuria AN. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access