Abstract
Background: Therapy-related Myelodysplastic Syndrome/Acute Myeloid Leukemia (t-MDS/AML) is a serious complication for cancer patients undergoing chemotherapy or radiation treatment. Determining the mechanisms behind disease progression will help to design targeted treatment strategies. The objective of this study was to investigate the genetic basis for t-MDS/AML development.
Case: Exome sequencing analysis was performed for a t-MDS/AML patient to investigate the somatic mutation in coding genes at diagnosis, remission, allo-transplantation, and relapse.
Results: The study observed dynamic changes of somatic mutation during disease development. A TGTG insertion mutation was indentified at exon 3 in Tet methylcytosine dioxygenase 2 (TET2) at diagnosis. This mutation caused a frameshift, generating a pre-matured stop codon downstream, and leading to loss-of-function of TET2. At post-allotransplantation and relapse, this mutation decreased to basal levels.
Conclusions: The results suggest that the TET2-mutated clone might play more important roles in initiation than in relapse of t-MDS/AML, in which a new clone(s) with wild-type TET2 may became dominant.
Keywords
Exome sequencing; Somatic mutation; tMDS/AML; TET2
Introduction
Chemotherapy and radiation in cancer patients can causea severe complication of therapy-related myelodysplastic syndrome/Acute myeloid leukemia (t-MDS/AML), a subgroup of MDS/AML disease [1,2]. Currently, successful preventative measures for this disease remain a challenge. The most effective treatment option is bone marrow allotransplantation with limited success [3,4]. Elucidating the genetic mechanism behind the development of t-MDS/AML is expected for developing new strategies for prevention and treatment of this disease.
Genetic factors are considered as the major cause for developing de novo MDS/AML, a more broad disease group compared to t-MDS/ AML. Studies have extensively tested for genetic abnormalities, which led to the identification of mutations in multiple functionally important genes, including TET2, TP53, DNMT3A, ASXL1, IDH1, and EZH2 [5,6]. Taking TET2 as an example: TET2 regulates DNA methylation though converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) [5]. Mutated TET2 fails to complete this conversion, can cause canceraffecting particularly the myeloid lineage [6,7]. Multiple studies revealed that somatic mutations in TET2 occur in de novo MDS/AML at high frequency [8-12]. A study in t-MDS/AML detected TET2 mutations in four out of 38 (10.5%) t-MDS/AML cases [13]. However, evidence is lacking to determine if the same genetic defficiencies in de novo MDS/ AML are responsible for t-MDS/AML. Further, why allotransplantation is not as effective in curing t-MDS/AML as in curing de novo MDS/AML remains unknown.
In the present study, we used exome sequencing [14] to analyze somatic mutations in coding genes in a t-MDS/AML patient at the disease stages of diagnosis, 69-days post-allotransplantation, 90-days postallotransplantation, and relapse. Comparison of the mutations identified during the different disease stages revealed their dynamic changes during disease development. Among the mutations included a TGTG-insertion mutation in TET2, a gene well-known for its roles in de novo MDS/AML. We observed its highly presence at diagnosis but diminished at postallotransplantation and relapse. This suggests that the TET2-mutated clone played important roles in initiating but not in relapse of the disease.
Case
Ethics statement
The subject involved in the study was informed using a questionaire addressing the following topics: reason behind study, purpose, study methods, risks, benefits to patient as well as others, alternatives to the study, renumeration, access to care at all times, protection of information, subject rights, and electing not to participate. The subject agreed to each of the questions, and signed in the consent form to participate in the study. The study was approved by the Institutional Review Board of University of Nebraska Medical Center under the protocol (488-11-FB).
Samples collection and DNA preparation
Samples were collected from the patient through the following: FFPE (Formalin Fixed Paraffin Embedded) fixed bone marrow cells at the diagnosis of t-MDS/AML, peripheral blood cells at pre-transplant (used only in Sanger sequencing validation), bone marrow samples at 69-days post allotransplantation, and bone marrow samples at 90-days post allotransplantation. A skin biopsy sample from the patient was collected before transplantation. A sample from peripheral blood cells of the bone marrow donor was also collected. Mononuclear cells from bone marrow and pripheral vein blood post allotransplantations were isolated by using Ficoll solution, and DNA was extracted from mononuclear cells using FlexiGene DNA kit (QiaGen, Coourtaboeuf, France). DNA from FFPE fixed cells at diagnosis and from skin biopsy were extracted using QIAamp kit following the manufacturer’s protocol.
Exome sequencing and mapping
Exome sequencing followed the standard Illumina exome procedure (Illumina, San Diego, CA, USA). The process included DNA fragmentation, adapter ligation, and exome fragment capturing using the TruSeq Exome Enrichment Kit v2 (Illumina). Sequences were collected in a Illumina HiSeq2000 sequencer using the paired-end mode (2X100). The following steps were used to process the exome sequences: (a) sequences were mapped to the human genome reference sequence human genome 19 (hg19) using Bowtie 2 sofware [15]; (b) resulting SAM files were converted to BAM files; (c) duplicates were removed using Picard (http://picard.sourceforge.net); (d) GATK-RealignerTargetCreator was used to complete local realignment [16]; (e) VarScan 2 was used to call variants [17]; (f) somatic single nucleotide polymorphisms (SNP) and indel variants were identified through filtering public databases including the Single Nucleotide Polymorphism Database 137 (dbSNP137), 1000 Genomes, Exome Variant Server 6500 (http://evs.gs.washington. edu/EVS/) (MAF>0.01). The variants from the patient samples were filtered with those from skin biopsy and donor cells to identify the somatic mutations present in cancer samples only. Mutations causing nonsynonymous, splicing change, and stop gain/loss were identified. Those causing damage effects were then predicted using ANNOVAR [18], which predicts deleterious effects by at least one of the six prediction programs of Sorting Intolerant From Tolerant (SIFT) [19], PolyPhen2 [20], LRT [21], MutationTaster [22], PhyloP [23], and GERP++ [24]. Sanger sequencing was used to validate the TET2 insert mutations.
Results
The patient analyzed in the study was a male Caucasian who at age 59 developed folicular lymphoma. Upon combinational chemotherapy with fludarabine, mitoxantrone, and dexamethasone, the patient achieved remission. He relapsed three years later and remissed upon treatment with rituximab. He relapsed again three years later and remissed again upon treatment with rituximab. Three year’s later, the patient developed thrombocytopenia, and had no response to prednisone. Bone marrow examination revealed hypercellular bone marrow (50%) with megakarryocytic hyperplasia and dyspoiesis, along with an increased number of blast cells (20%). Cytogenetic analysis revealed the presence of two karyotype lines of 46, XY in 17 out of 21 metaphases and complex karyotype of 42-44, add(X)(q13),del(2)(p21),add(4)(q31),del(5) (q13q35),-6,-7,add(10)(q26),-18+1-2mar[8]/46,XY[12] in 4 out of 21 metaphases. The patient was diagnosed as t-MDS/AML. Upon two-cycle treatment of decitabibe, myeloblast cells in bone marrow decreased to 2%. Subsequently, the patient received bone marrow allotransplantation following standard care from the Stem Cell Transplant Center at the University of Nebraska Medical Center. Further, the patient received combinational treatment of fludarabine and busulfan. Bone marrow examination at 90-days post allotransplantation showed 5% blasts. Flowcytometry with 15-CD markers showed residual myelodysplasia. Cytogenetic analysis showed normal karyotype of 46 XY. MDS-specific FISH analysis [del5q31, KMT2A (MLL; 11q23) locus rearrangement, trisomy 8, monosomy 7, deletions of 7q31, 20q12 and 20q13] showed no sign of MDS-related abnormalities. The patient died three months later, at age 69.
DNA was extracted from bone marrow cells of the patient at the time of t-MDS/AML diagnosis, 69-days post-allotransplantation, and 90-days post-allotransplantation. DNA was also extracted from skin of the patient and from the peripheral blood cells of the allotransplant donor. DNA samples were used for exome sequencing. Genomic variants were called from each sample. Variants of normal polymorphisms were removed by filtering through dbSNP, 6500 Exomes, and 1000 Genome databases with a minor allele frequency (MAF)>0.01 as the cut-off. Further, the variants shared with those from patient’s skin, and the variants in posttransplantation samples shared with donor’s sample, were removed. From the remaining vaiants, nonsynonymous deleterious single nucleotide variation (SNV) and indels were identified at different disease stages: six mutations at diagnosis, 15 mutations at 69-days post-allotransplantation, and 20 mutations at 90-days post-allotransplantation (Tables 1 and 2). The mutations between the three stages were compared and showed that no mutations were shared between diagnosis and post-transplantation; eight mutations were shared between 69-days and 90-days post-transplantation (Figure 1).
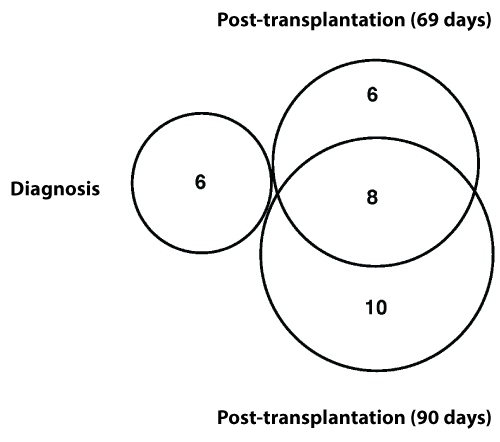
Figure 1: Dynamic changes in somatic mutations during disease development-Somatic mutations identified at diagnosis, relapse 69 days post-transplantation and 90 days post-transplantation were compared. No mutations were shared between diagnosis and post transplantations, and 8 mutations were shared between 69 days and 90 days post transplantation.
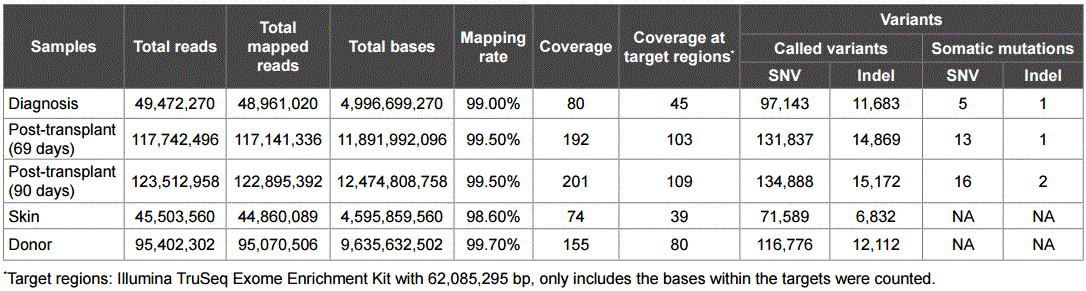
Table 1: Exome sequencing and mapping results.
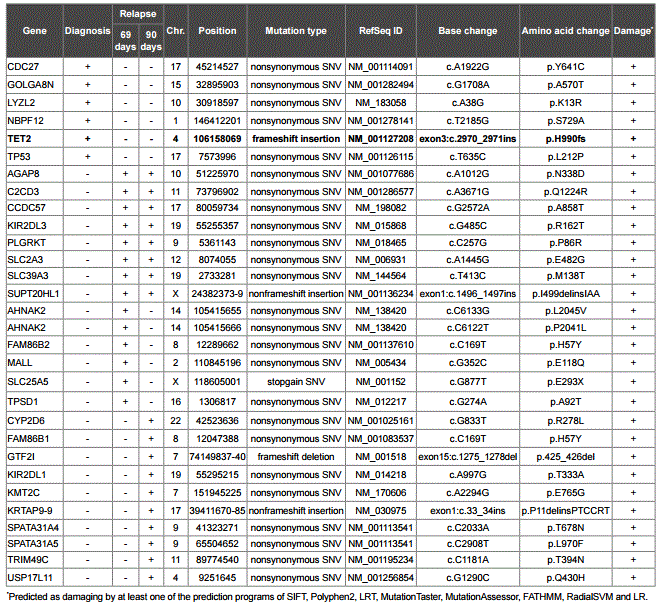
Table 2: Somatic mutations identified in the t-MDS/AML case.
Among the mutations detected, a four base insertion (TGTG) mutation at exon 3 of TET2 was identified at diagnosis but not post-transplantation. Of the 2,002 amino acid residues of TET2 (RefSeq NP_001120680.1), the insertion mutation at 990 caused a frameshift, generated a stop codon TAA at 1009, and led to the loss-of-function of TET2 (Figure 2A). Comparing with the mutations identified in the t-MDS/AML study [13] showed that this mutation is a novel mutation in t-MDS/AML (Figure 2B).
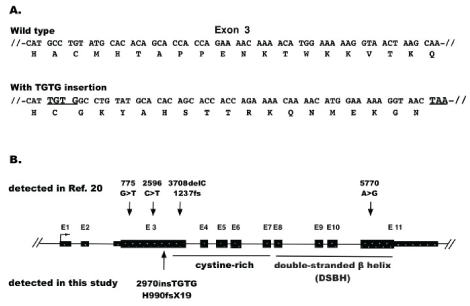
Figure 2: TET2 mutation. A) TGTG-insertion mutation. It is located at exon 3, causing a coding frame shift and creating a pre-terminate TAA codon downstream. B) Positions of mutations in TET2 in t-MDS/AML. The four mutations marked at the top level were identified by the study [20], the mutation marked at the bottom level was identified by this study
Sanger sequencing was used to validate the TET2 mutation at different stages of disease development. The mutation was confirmed to be a true somatic mutation. It was present at diagnosis, absent before transplantation, and reappeared post-transplantation at relapse, albeit at very low levels that was under the threshield of variant calling by the standard exome mapping conditions (Figure 3). To provide quantitative information for the TET2 mutation, the ratio in Sanger sequence signals was compared between the wild-type GCCT and the insert TGTG at the same positions in the samples of diagnosis, 69-days post allotransplantation, and 90-days post allotransplantation. The results showed that the TGTG insert signal reached 35% of the wild-type GCCT signal at diagnosis, but the rates decreased to 9% at 69-days and 4% at 90-days post allotransplantation (Table 3). The number of wild-type GCCT sequences and insert TGTG sequences in the exome data were also counted, showing that the mutated sequences accounted for 14% (5/35) at diagnosis, 6% (4/64) at 69-days, and 6% (3/51) at 90-days post-allotransplantation. The two sets of quantitative results indicated that the mutated TET2 sequences were at much higher levels at diagnosis, but diminished at relapse and post allotransplantation.
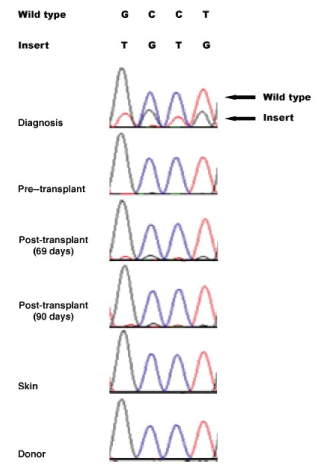
Figure 3: Dynamic presence of TET2 TGTG insertion at different disease stages revealed by Sanger sequencing-The TET2 TGTG insertion was highly present at diagnosis, absent pre-transplantation, and appeared again at post-transplantation and relapse but at basal levels. The same mutation was absent in patient’s skin and donor’s cells.
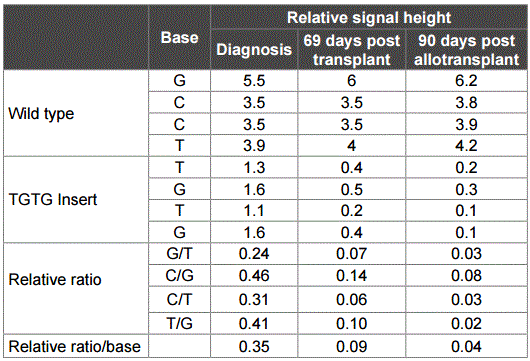
Table 3: Quantitative relationship between wild type and insert signals in TET2.
Discussion
Precision medicine has been proposed to be a new model of medicine. By following each patient’s disease process at genomic and molecular level, precision medicine provides hope to identify specific genetic markers for targeted treatment. Therefore, understanding the genetic changes during disease development is important to reach this goal. Instead of testing mutations once in a patient during disease, our study analyzed the somatic mutations in a t-MDS/AML patient across the entire disease development to trace the fate of somatic mutation during disease development. The results revealed dynamic changes of multiple somatic mutations as best examplified by the mutation in TET2, a gene known to play important roles in de novo MDS/AML [5-13].
As part of treatment for t-MDS/AML, successful allotransplantation is expected to replace the hematopoietic system of the patient with that of the bone marrow donor, allowing the patient to regain normal hematopoietic function. However, the relapse of the patient post transplantation implies that this did not happen, or happened transiently such that the hematopoietic system of the patient gained dominance and was responsible for the relapse. However, the clone(s) at the time of relapse was unlikely the same as the one at the time of diagnosis, as evidenced by (1) very different mutation pools at diagnosis and relapse (Figure 1), and (2) higher-level the mutated TET2 at diagnosis but lower at the relapse (Figure 3 and Table 3), which was concurrent with lower blast rate at the relapse. It is possible that at the time of the last biopsy 90-days post transplantation, the TET2-mutated clone was still rare but became dominant afterwards. However, this possibility was not supported by the cytogenetic and FISH data at 90-days post-allotransplantation, which showed a normal karyotype and no sign for MDS-specific abnormalities. Our explanation is that the TET2-mutated clone contributed significantly in initiation but not in relapse of the t-MDS/AML patient.
Conclusions
The results from our study indicate that following dynamic change of somatic mutations in disease-specific genes allows more precise monitoring genetic basis of disease develoopment and will help selection of personized treatment for patients.
Data Deposition and Access
The exome data were deposited in European Nucleotide Archive (ENA) with accession ID: PRJEB18718
Conflict of Interest
Authors declare no competing interests in the study.
Author Contributions
PC performed the experiments, YK performed bioinformatics data analysis, MA identified and provided patient materials, MA, SMW conceived the study, SMW designed the study, and wrote the manuscript.
Acknowledgments
The study was supported by a grant from Fred & Pamela Buffett Cancer Center, University of Nebraska Medical Center (MA) and a pilot grant from Fred & Pamela Buffett Cancer Center, University of Nebraska Medical Center (SMW).
Article Information
Article Type: Case Report
Citation: Chen P, Kim YC, Akhtari M, Wang SM (2017) An Example of using Precision Medicine in Cancer Care: Dynamic Change of TET2 Mutation in a Patient with Therapy-related Myelodysplastic Syndrome/ Acute Myeloid Leukemia. Int J Cancer Res Mol Mech 3(2): doi http://dx.doi.org/10.16966/2381-3318.134
Copyright: © 2017 Chen P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 22 May 2017
Accepted date: 26 Jun 2017
Published date: 30 Jun 2017