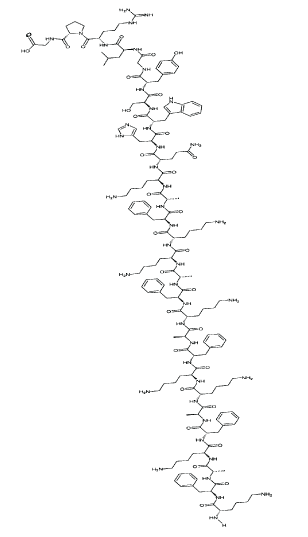
Figure 1: Structural Formula of EP-100.
Leuschner C1* Coulter A2 Keener J3 Alila H1
1Esperance Pharmaceuticals, Houston, Texas, United States*Corresponding author: Leuschner C, Esperance Pharmaceuticals, 909 Fannin Street, Suite 2100, Houston, Texas 77010, United States, E-mail: Carola@esperancepharma.com
Article Type: Research Article
Citation: Leuschner C, Coulter A, Keener J, Alila H (2017) Targeted Oncolytic Peptide for Treatment of Ovarian Cancers. Int J Cancer Res Mol Mech 3(1): doi http://dx.doi.org/10.16966/2381-3318.132
Copyright: © 2017 Leuschner C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Aim: EP-100 is a novel, LHRH receptor targeted cytolytic peptide conjugate designed to specifically kill LHRH receptor positive cancer cells. This paper presents studies that characterize EP-100, including specificity of binding to the LHRH receptor, mechanism of action, screening of cancer cell lines, correlation of cell line sensitivity to LHRH receptors expression levels and In vivo efficacy.
Methods: Binding kinetics of EP-100 to the LHRH receptor were evaluated in a saturation binding study. Immunohistochemistry and confocal microscopy after labeling with fluorescent LHRH conjugates were used to detect LHRH receptors on human cancer cell lines and tumor tissues. in vitro cytotoxicity screening in human cancer cell lines, including ovary, breast, endometrial, prostate, pancreatic and uterine sarcoma cell lines, were assessed through cell viability assays. In vivo efficacy studies were conducted by systemically injecting EP-100 into ovarian cancer xenograft bearing mice. Treatment efficacy was assessed through histological evaluation, tumor marker CA-125 measurements and PET imaging.
Results: These EP-100 studies demonstrated high affinity binding of EP-100 to the LHRH receptor, cytotoxicity and specificity of EP-100 in human cancer cell lines in vitro at low micromolar levels. EP-100 rapidly destroys LHRH receptor positive cancer cells through membrane disruption within hours. In vivo studies in single and repeat dosing demonstrated tumor regression of ovarian cancer xenografts at doses of 0.2 mg/kg of EP-100, safety and absence of effect on vital organs and red blood cells. Untargeted lytic peptide was without effect in vitro and In vivo. EP-100 killed tumors by necrosis. Tumor death was confirmed by PET imaging and tumor marker (CA-125) reduction in each treatment group.
Conclusions: The studies on EP-100 on cancer cells in vitro and In vivo demonstrate the characteristics and mechanism of action of a newly designed targeted synthetic, cationic peptide with remarkable potency to destroy specifically LHRH receptor expressing ovarian cancer cells through lysis of the cancer cell membrane leading to necrosis. EP-100 represents an appropriate candidate for treatment of ovarian cancer.
Ovarian cancer; Lytic Peptides; Targeted therapy; Targeted lytic peptide; LHRH; LHRH receptors
LHRH: Luteinizing hormone releasing hormone; LH: Luteinizing hormone; FSH: Follicle stimulating hormone; PET: Positronemission tomography; CT: Computed tomography
Ovarian cancer is the fifth leading cause of cancer deaths with 14,080 cases (5%) while 195,000 women still living with the disease [1]. Seventy percent of the 22,000 women diagnosed annually with ovarian cancer succumb to death, despite tumor ablation and rigorous chemotherapeutic intervention. Often, diagnosis occurs at an advanced stage, because of unspecific symptoms and lack of effective screening methods. Only 27% of patients with ovarian cancer survive 5 years if diagnosed with distant metastatic disease [1].
The standard of care (SOC) treatments for ovarian cancer, after debulking surgery, are generic platinum agents, paclitaxel, etoposide, gemcitabine, liposomal doxorubicin (Doxil®/Caelyx), and topotecan [2], generating low response rates of less than 18 months [3]. In 2014, bevacizumab (Avastin®) received FDA Fast Track approval for recurrent platinum-refractory patients despite the inability of its progression free survival (PFS) benefit to translate to overall survival (OS) benefit [4]. Recently, targeted therapeutics such as a PARP-inhibitor demonstrated responses in a subgroup of ovarian cancer patients that showed BRCA mutations [5,6]. Systemic treatments with untargeted compounds (Carboplatin, Taxol, Doxil) cause serious side effects, and do not discriminate between cancer and normal cells in vital organs. They are designed to kill rapidly dividing cells and are ineffective in killing non dividing metastatic cancer cells, the major cause of cancer related deaths. Patients’ tumors that develop multi-drug resistance often present with more aggressive and deadly forms of the disease. New drugs with novel mechanisms of action that specifically target cancer cells are needed to treat patients that have no other options.
One of the most studied targets of cancer cells is the luteinizing hormone releasing (LHRH) receptor. A number of most frequently occurring cancers over-express receptors for LHRH; these include ovarian cancers (80%), breast (52 %), endometrial (80%), prostate (80%), kidney, pancreatic (62%), colon cancers and hematological malignancies (94%) [7-10].
Consequently, the delivery of toxins through LHRH conjugation continues to be intensively studied, and proof of concept and rationale for LHRH receptor targeting have been established. Generally, LHRH receptors are present on the surface membrane and are overexpressed on cancer cells, but not in normal tissues except for pituitary glands and gonads [11,12]. Several studies reported pre-clinical proof of concept for LHRH-toxin conjugates such as bovine RNaseA [13] pokeweed protein [14] and Pseudomonas exotoxin [15], proteins involved in apoptosis, such as BIK, BAK, BAX and DFF40 [16]. Chemotherapeutics such as doxorubicin [10], paclitaxel [17], melphalan, cisplatin and methotrexate have been conjugated to LHRH [18,19]. A conjugate of doxorubicin linked to an LHRH agonist (AEZS-108, Zoptarelin doxorubicin, Aeterna Zentaris) is currently in Phase III clinical trials for endometrial cancers [10]. Zoptarelin doxorubicin and other LHRH toxin conjugates require elaborate chemical synthesis, and involve optimized linker technologies to release the toxin inside the target cells to exert activity.
An alternative approach for killing cancer cells has been developed using targeted lytic peptides that have a unique and novel mechanism of action. Lytic peptides occur abundantly in nature, and serve as defense molecules for bacteria, plants, insects, invertebrates, vertebrates and humans [20-23]. They consist of 9-60 amino acids, can be anionic or cationic, linear or cyclic, and share an amphipathic structure that allows them to interact and intercalate with cell membrane structures of opposite charge [20-22]. Examples of alpha helical cationic lytic peptides are cecropins and melittin from insects, magainin, dermaseptin from amphibians, cathelicidin, granulysin, perforin, defensin4 from humans. All these lytic peptides interact with negatively charged membranes of bacteria or cancer cells, and cause cell death through membrane lysis. Natural lytic peptides lack cell selectivity and their hemolytic activity and antigenicity limits their use for clinical applications. Intensive studies on lytic peptide design generated synthetic lytic peptides consisting of L and D amino acids or amino acid analogues to increase stability. These efforts produced synthetic lytic peptides with increased activity against cancer cells, and reduced hemolytic activity and lack of immunogenicity [24-28]. They have generally an alpha helical cationic structure that lysis negatively charged membranes rapidly. The mechanism of action for killing cells varies and is dependent on the composition and sequence of the lytic peptide. Lytic peptides that kill by necrosis are generally not internalized. Others maybe internalized and have been shown to elicit apoptosis [23]. Because these lytic peptides were not targeted, their in vivo applications were limited to injections directly into the tumors where necrosis was observed in treated animals as well as immune cell infiltration. To date, only one untargeted lytic peptide has been tested in clinical trials. The lactoferricin derived synthetic lytic peptide, LTX-315, has been tested in clinical trials in solid tumors using intratumoral injections [27-30].
Further improvement of lytic peptide activities has been achieved by conjugation to targeting ligands or antibodies for which receptors are over expressed on cancer cells [31-40]. This approach not only improved anticancer activity but also allowed systemic injection for generating activities against primary tumors and metastases in various xenograft mouse models [34,41-49].
The first selective cancer-targeting peptides consisted of synthetic lytic peptides (Phor14, Hecate and later, Phor21) that were conjugated to peptides to target human chorionic gonadotropin (hCG) receptor and LHRH receptor [42-49]. These compounds rapidly and specifically killed cells over-expressing hCG or LHRH receptors. The direct contact and interaction with the cell membrane caused cell membrane disruption and death by necrosis. Experiments in animal models of prostate, breast and ovarian cancers showed that these compounds regressed established tumors following three weekly intravenous injections at doses of 10 mg/kg for Hecate and Phor14 conjugates, and at doses as low as 0.2 mg/kg for Phor- 21-βCG [33,43-47]. These conjugates did not cause systemic toxicities, were not antigenic and had high potency even while having a short halflife of less than 10 minutes [50]. They also killed multi-drug resistant cancer cells, an advantage over existing cancer chemotherapeutics. Unlike other targeted small molecules or antibody drug conjugates currently under development [9,17,51,52], the lytic peptide conjugates kill cells on contact and do not need to be internalized or cleaved to gain activity against the target cells.
For clinical development, a LHRH receptor targeted novel, more potent cytolytic peptide conjugate was designed, synthesized and tested to increase toxicity and specificity to cancer cells without harming red blood cells. These studies resulted in the development of EP-100 for targeting LHRH receptors on cancers cells. EP-100 is a 28 amino acid peptide (MW=3,317 g/mol) comprised of a natural hormone sequence of the luteinizing hormone releasing hormone, joined to an 18 amino acid lytic peptide (called CLIP71) without a linker (Figure 1). EP-100 targets and specifically kills cancer cells that over-express LHRH receptors via a novel mechanism of action that involves direct membrane disruption and necrosis. The lytic domain, CLIP71, is a linear, alpha helical, cationic, amphipathic membrane-disrupting peptide. Pre-clinical studies demonstrated cytotoxicity towards LHRH receptor expressing cell lines, and demonstrated synergy with standard of care anti-cancer drugs and reversed drug resistance [53-55]. These favorable efficacy and safety features of EP-100 prompted further development for clinical trials. Two clinical trials were conducted with EP-100 as single agent in patients with solid tumors (NCT0094955) and EP-100 in combination with paclitaxel in advanced ovarian cancer patients (NCT01485848) [56,57]. A remarkable safety and tolerability profile was observed after a total of 87 patients treated for up to 18 months.
Figure 1: Structural Formula of EP-100.
This paper reports pre-clinical data that were conducted prior to the clinical studies. Most of the data were included in the data collection for the IND. Since then, additional data have been generated and were included in this pre-clinical publication.
In this paper, pre-clinical studies on EP-100 are presented that include studies on the mechanism of action, specificity of binding to the LHRH receptor, binding kinetics and in vitro cytotoxicity studies in human cancer cell lines of solid tumors (ovary, breast, endometrial, prostate, pancreatic, melanoma and uterine sarcoma) to determine potential indications. The mechanism of action and pharmacokinetics were studied in vitro. The binding parameters of EP-100 to the LHRH receptor were determined. The specificity and potency of EP-100 was assessed in relation to LHRH receptors on 18 cancer cell lines from solid human tumors. As an example for in vivo efficacy studies, results from ovarian cancer xenografts were included.
Cell lines were obtained from American Type Cell Culture Collection, Manassas, VA. EP-100, CLIP71 and FITC-D-Lys6 -LHRH were synthesized by standard solid-phase chemistry methods with Fmoc [Nα- (9-Fluorenylmethoxycarbonyl)] and purified by standard reverse-phase high pressure liquid chromatography (HPLC) by American Peptide Company, Sunnyvale, CA, and provided in lyophylized form. The method for synthesizing EP-100 is proprietary to American Peptide Company.
Binding kinetics of EP-100 to the LHRH receptor were evaluated in a saturation binding study. The assay was conducted as described by Leuschner et al. using a binding buffer consisting of 0.3M sucrose, 1 mM EDTA, 0.025 mM PMSF, 1 mM DTT, 2 µg/ml aprotinin and 25 mM Tris, pH 7.5 containing 0.2% BSA [46]. Vials were coated overnight with incubation buffer containing 2.5% BSA. Specific binding was determined using 30 µg of a membrane preparation (Millipore ChemiScreen™ human GnRH receptor I, HTS027M, lot number JH1424114, Millipore, Inc, Billerica, MA) and a membrane preparation of the uterine sarcoma cell line MES-SA-Dx5. Membranes were prepared as described in Leuschner et al.. Binding was conducted using 75,000 cpm of 125-I-D-Trp6 -LHRH (NEX 365010UC, lot # IZ41230, Perkin Elmer) in the presence of increasing concentrations of the LHRH analogues Triptorelin (APC 54- 1-21, lot # R02030A1) or leuprolide (Sigma L0399, lot # SLBC4150V) and compared to EP-100 (APC338613, lot # W011007C1). Non-specific binding was determined using a 1000x unlabeled compounds. Membranes in reaction mixture (100 µl) were incubated for 90 minutes on ice and the reaction terminated by adding 1 ml of ice cold binding buffer and centrifuged at 19,500xg and washed three times with binding buffer. The bound radiolabel was determined using a gamma counter. Binding was calculated using Graph Pad Prizm 5™ analysis program for saturation binding kinetics.
Membrane action of EP-100 on cancer cell lines was determined using confocal fluorescence microscopy using 2 different methods. First, human melanoma cells (MDA-MB-435S) were seeded at a density of 10,000 cells onto culture dishes and nuclei and mitochondria were stained using fluorescent dyes (DRAQ5™ (Alexis Corporation, USA) – blue and MitoTracker® Red CMXRos (M7512) (Molecular Probes, Inc. OR, USA). Cell membranes were stained with wheat germ Alexa Fluor green conjugates (Molecular Probes, Inc. OR, USA).
EP-100 reconstituted in saline was added at a final concentration of 10 µM and incubated for 5-60 minutes. Culture dishes with saline only served as controls.
MDA-MB-435S and Skov-3, were grown to 80% confluence on glass cover slips coated with poly L- lysine. Cells were stained with 1 µM Mitotracker Red CMXRos for visualization of mitochondria (InVitrogen lot# 513724) and 500 nM DAPI for visualization of nuclei (InVitrogen lot# 493001) in growth medium for 30 min at 37°C. Either saline (untreated control) or 2 µM FITC- labeled EP-100 (APC lot# W10051C1) was added to cells at 37°C for time periods of 2 to 30 minutes. Polarization/ depolarization of mitochondria was studied using the fluorescent redox marker JC-1 at 5 µM in the presence of 5 µM EP-100 or saline and compared to the mitochondrial depolarizing agent, the protonophore m-chlorophenylhydrazone CCCP (1 mM). Cells were fixed in 4% paraformaldehyde dissolved in growth medium. Imaging was performed using a Zeiss confocal microscope with a 3I Everest imaging system at 40X and 63X magnification.
Cells were seeded at a density of 10,000 cells/well into chamber slides. Confluent monolayers were fixed in 10% phosphate buffered formalin and preserved in ethanol after successive dehydration. Immunoperoxidase staining using the mouse monoclonal antibody for LHRH receptors (GNRH03, #MS-1139-P, LabVision) and analyzed on Ventana XT, Ventana Benchmark or Ventana Nexes Units. Image analysis was conducted with the Ventana Image Analysis System (VIAS) adjunctive computer assisted image analysis system functionally connected to an interactive microscope (Axio Imager). The quantitative analysis was conducted with the program for quantification of Her2/neu receptor that included morphometric and colorimetric analysis. LHRH receptor status results were reported as percentage of cells showing positive membrane staining under the following criteria: 0 non-immunoreactive, 1+: 1-25% positive, 2+26 – 50% positive, 3+51-75% positive cells. These data were compared to manual assessments of strength of staining.
FITC-D-Lys6 -LHRH (APC 365254, lot 1302084C) was used for detection of surface LHRH receptors on formalin fixed monolayers at a concentration of 2 µM, FITC incubations served as controls. Nuclei were stained with DAPI.
RNA was isolated from cell cultures using the RNeasy Mini Kit (Qiagen #74104, source), quantified and analyzed for integrity using an Agilent 2100 Bioanalyzer. Total RNA from the ovarian cancer cell line, OVCAR-3, was used for the standard curve. Primer Express software (Applied Biosystems) was used to design Taqman PCR primers and probe matching nucleotides encoding amino acids 2-24 in the LHRH-R, a highly conserved region involved in hormone binding.
Reverse transcriptase and PCR were performed in a one step reaction with the reverse primer specifically priming synthesis of LHRH-R cDNA in the first stage. The same reaction mixture was used for all standards and samples, and each sample was assayed in triplicate. Real-time RTPCR reactions were conducted in 96-well optical plates (ABI #4306737) with MicroAmp Optical Adhesive Film (ABI #4311971) in an ABI 7900 Sequence Detection System with the first reaction stage set to 48°C for 30 minutes for the cDNA synthesis step. Fifty ng of cellular RNA was used for each reaction. Target gene mRNA levels were normalized based on Agilent quantification of sample total RNA concentration.
In vitro screening: Human cancer cell lines included: ovarian cancer cell lines (OVCAR-3, SKOV-3, A2780), breast cancer cell lines (MDAMB-231, BT474, SKBR-3, MCF-7, T47D), endometrial cancer cell lines (Hec1A and An3-CA), prostate cancer cell lines (PC-3, LnCaP), pancreatic cancer cell lines (Panc-1, BxPC-3, MiaPaCa), a kidney cancer cell line A498, a melanoma cell line (MDA-MB-435S) and a multi-drug resistant clone of the human uterine sarcoma cell line, Mes-SA-Dx5. LHRH receptor negative cell lines such as the human ovarian cancer cell line SKOV-3, and two non-cancerous epithelial cell lines, the mouse fibroblast cell line 3T3 and the human breast epithelial cell line MCF-10A served as negative controls for the target receptor. Cells were seeded in 96 well plates using 2,000-10,000 cells/well. After 48 hours EP-100 or unconjugated lytic peptide (CLIP71) dissolved in saline were added to the multi-well plates at increasing concentrations of 0, 0.001, 0.01, 0.1, 1, 2, 5, 10 and 100 µM. Cells were incubated for 4 or 24 h at 37° C. Kinetics of activity were determined as described above by following cell viability after 15 and 30 minutes and 1, 2, 4, and 24 hours. Cell viability was determined using formazan conversion assays (MTT assays) or luminometric cell viability assays (Cell-Titer-Glo, Promega, Madison, WI). Membrane integrity was measured using a luminometric assay for dead cell protease (Cytotox-Glo, Promega, Madison, WI). Controls contained USP saline or 0.1% TritonX-100™ as reference for 0 and 100% cell death, respectively. Data were processed and analyzed using Graph Pad Prizm 4™ software and Graph Pad Prizm 5™ (Graph Pad Prizm, Inc). Statistical analysis for significance was determined by a two-tailed Student’s T-test.
In vivo studies: In vivo studies were approved by the Institutional Care and Use Committee, Pennington Biomedical Research Center, Baton Rouge, LA, Protocol number 372 and 543P.
Tumors were induced in female nude mice (Balbc/NuNu, 5 weeks old) by SC injection of 4x106 NIH:OVCAR-3 cell suspension in Matrigel™ (passage # 34). EP-100 (APC 338913, Lot no. V09108X1) was freshly reconstituted in saline and administered IV at a dose of 0.02, 0.2, or 2 mg/ kg on Days 33, 41, and 47 following tumor cell inoculation. Cisplatinum was administered IP to a separate group of mice at a dose of 3 mg/kg on Days 33, 34 and 35. Control groups received saline or unconjugated lytic peptide CLIP-71 (2mg/kg). A group of untreated tumor- bearing mice was sacrificed on Day 33 and served as a baseline control. All other mice were sacrificed on Day 51 or 52 for assessment of tumors and serum levels of the tumor marker CA-125. CA-125 was determined in serum, collected from each individual mouse at necropsy using a Enzyme Linked Immunoassay for quantitative determination of ovarian cancer antigen CA-125 (Assay kit Genway, Biotech, Inc. San Diego, CA, Catalog #40-052-115009, #BC- 1013 according to the manufacturer).
Xenografts were propagated SC from NIH:OVCAR-3 tumor cells (passage number 48) as Matrigel™ suspension into female nude mice (Balbc/NuNu, 5 weeks old). EP-100 (APC 338913, lot # P080401) was reconstituted in saline at a dose concentration of 0.04 mg/ml for lateral tail vein injection. Treatment at a dose of 0.2 mg/kg (N=16) started on day 38 after tumor cell injection and continued on days 41, 44, 48, 52, 59 and 61. Saline injected mice served as controls (N=13). Tumor volumes and body weights were recorded. On day 85 after tumor inoculation, mice were sacrificed and tumors weighed and fixed in formalin. On day 78, four mice of saline control and the EP-100 treatment group were prepared for tumor assessment through PET-CT imaging. Overnight fasted mice were injected with 0.2 µCi [18F]-2-fluoro-2-deoxy-D-glucose (FDG) in a volume of 0.1 ml into the lateral tail vein. PET/CT imaging was conducted after 1 hour on sacrificed mice.
Statistical analysis of data was conducted using the two-tailed student’s T-test, one way analysis of variance (ANOVA), two tailed Student’s T-test and Tukey-Kramer multiple comparison tests to determine significance among means. Analyses were calculated using Graph Pad Prizm 5™ and Instat 3 (Graph Pad Prizm, Inc).
EP-100 is a 28 amino acid lytic peptide conjugate that consists of the lytic portion CLIP71 (18 amino acids) conjugated to LHRH and has the molecular formula C163H243N43O32 and a molecular weight of 3317.3 Daltons. Figure 1 shows the structural formula of EP-100. It is producing a white to off-white lyophilized substance and readily dissolves in water or hydrophilic buffers.
Binding kinetics were determined in saturation binding experiments for EP-100 in comparison with two LHRH analogues, Triptorelin and Leuprolide. The LHRH analogue, Triptorelin, was used, because of its increased stability due to D-Trp on position 6. Moreover, Triptorelin showed similar specific binding to the LHRH receptor as the natural LHRH [58]. The binding parameters for Chem1-LHRH RI transfected cell membranes and Mes-SA-Dx5 cell membranes are summarized in Table 1. Binding parameters for affinity (KD) and capacities (Bmax) to the LHRH receptor were similar for Triptorelin, Leuprolide and EP-100, all of which displaced radiolabeled Triptorelin on membranes of Chem 1-LHRH R I transfected cells (Figure 2A). Binding capacities (Bmax) and affinities (KD) were determined using Scatchard analysis. One binding site was identified with KD values of 2.2±0.2 nM, Bmax 1,538±214 fmol/mg protein for Triptorelin, 2.1±0.1 nM, Bmax 1,395±282 fmol/mg protein for Leuprolide. Binding parameters were similar for EP-100 with KD 2.9±0.1 nM and Bmax of 1,358±337 fmol/mg protein. The binding parameters on membranes from a uterine sarcoma cell line (Mes-SA-Dx5) showed a single binding site with affinity to the LHRH receptor of KD of 3.7±0.4 nM for Triptorelin and 4.1±0.3 nM for EP-100 and binding capacities (Bmax) of 1,600±256 and 1,851±281 fmol/mg protein (Figure 2B). EP-100 bound to LHRH receptors with high affinity and displaced radiolabeled Triptorelin in a dose dependent fashion. The binding potential (Bmax/KD) for individual compounds were comparable for Chem-1 membranes with 686.6 and 635.1 for Triptorelin and Leuprolide, and 477.6 for EP-100. On Mes-SA-Dx5 membranes binding potential was 427.8 for Triptorelin and 451.5 for EP-100. The data confirmed that EP-100 is specifically binding the LHRH receptors with high affinity.

Figure 2: A) Saturation binding curve of I125-D-Trp6 -LHRH on cell membranes from a LHRH receptor I transfected Chem 1 cell line with Scatchard plot inset. Triptorelin: KD 2.2±0.2 nM, Bmax 1,538±214 fmol/mg protein, Leuprolide: KD 2.1±0.1 nM, Bmax 1,395±282 fmol/mg protein and EP-100: KD 2.9±0.1 nM, Bmax 1,358±337 fmol/mg protein. B) Saturation binding curve of I125-D-Trp6 -LHRH on cell membranes from a uterine sarcoma cell line (Mes-SA-Dx5) with Scatchard plot inset. Triptorelin: KD 3.7±0.4 nM, Bmax 1,600±256 fmol/mg protein, and EP-100: KD 4.1±0.3 nM, Bmax 1,851±281 fmol/mg protein.
| Binding Parameter | EP-100 | Triptorelin | Leuprolide |
| Chem1-LHRH RI | |||
| KD [nM] | 2.9±0.1 | 2.24±0.2 | 2.14±0.1 |
| Bmax [fmol/ mg protein] | 1385±337 | 1538±214 | 1395±282 |
| Mes-SA-Dx5 | |||
| KD [nM] | 4.09±0.3 | 3.74 | Not determined |
| B max [fmol/mg protein] | 1851±281 | 1600±256 | Not determined |
Table 1: Binding parameters of EP-100 and LHRH analogues on Chem1- LHRH RI and Mes-SA-Dx5 membrane preparations calculated from Scatchard analyses.
LHRH receptors on 3 ovarian cancer cell lines were visualized in confocal microscopy images using FITC-D-Lys6 -LHRH for LHRH receptors and FITC staining as controls. The upper panel for Figure 3 shows background staining from FITC incubated monolayers that are similar for all three cell lines. The lower panel of Figure 3 shows the surface staining as green color in the ovarian cancer cell lines OVCAR-3 and A2780. The ovarian cancer cell line SKOV-3 shows only the nuclear staining (blue) whereas the LHRH receptor was undetectable as seen by absence of the green label that represents LHRH receptors.

Figure 3: Detection of LHRH receptors on cancer cell surfaces in confocal microscopy through binding of D-Lys-FITC-LHRH. Fixed monolayers of ovarian cancer cell lines were incubated with 2 µM DLys-FITC-LHRH or FITC for 30 minutes and DAPI for nuclear staining. FITC control stains showed only background label and was similar for SKOV-3, OVCAR-3 and A2780 cell lines. Green FITC label (positive for LHRH receptors) was present in the two ovarian cancer cell lines OVCAR-3 and A2780. The ovarian cancer cell line SKOV-3 tested negative for LHRH receptors.
Screening of 18 human cancer cell lines from solid tumors of uterine sarcoma (1), breast (5), prostate (2), ovarian (3), endometrial (2), kidney (1), melanoma (1) and pancreatic (3) cancers resulted in maximal potency measured as IC50 values for EP-100 at low micromolar levels ranging from 0.6-6.6 µM after 4 hours with no further decrease after 24 hours (Table 2). The mouse fibroblast cell line 3T3 and the breast epithelial cell line MCF- 10A were not killed after 4 or 24 hours by either EP-100 or CLIP71. In contrast, activities for unconjugated CLIP71 ranged from 13.7-181 µM after 4 hours and 11.2-109 µM after 24 hours or showed no toxicity at all. The PC-3 prostate and the BT-474 breast cancer cell line were not killed by CLIP71 after 4 hours. The LHRH receptor negative human ovarian cancer cell line SKOV-3 showed similar sensitivities for EP-100 and CLIP71 with IC50 values of 50.5±5.3 resp 49.2±3.8 µM after 4 hours and 21.1±0.9 resp 28.2±4.9 µM after 24 hours. EP-100 was effective against multi-drug resistant cancer cell lines that expressed the LHRH receptor such as MES-SA-Dx5 (0.58±0.009 µM, 230.62±0.009 µM), OVCAR-3 (5.8±0.3 µM, 3.0±0.5 µM) and BT-474 (6.6±0.03 µM, 0.9±0.02 µM) at low micromolar levels, but not to LHRH receptor negative SKOV-3 cells. The non-cancerous mouse fibroblast cell line NIH-3T3 (negative for LHRH receptors) was not killed after 4 hours by either EP-100 or CLIP71 and showed similar sensitivities after 24 hours for both compounds of 121.2±16.5 respectively 102.7±6.8 µM. The human breast epithelial cell line MCF-10A was resistant to EP-100 and CLIP71 with IC50 values 15.3±2.5 µM and 22.6±2.5 µM after 24 hours (Table 2). Unconjugated lytic peptide, CLIP71, was more than 20- fold less toxic to cancer cell lines with IC50 values ranging from 13.7-181 µM after 4 hours and 11.2-109 µM after 24 hours (Table 2). For LHRH receptor positive cell lines the full efficacy was reached after 4 hours and did not further increase when cell death was measured after 24 hours in LHRH receptor positive cell lines.
| Cell line | IC50 [µM] 4 h EP-100 | IC50 [µM] 24 h EP-100 | IC50 [µM] 4 h Clip-71 | IC50 [µM] 24 h Clip-71 |
| Uterine Sarcoma | ||||
| MES-SA-Dx5 | 0.58±0.009 | 0.62±0.01 | 13.75±0.16 | 11.2±1.1 |
| Melanoma | ||||
| MDA-MB-435 | 0.59±0.001 | 0.62±0.002 | 38.02±8.5 | 27.4±3.1 |
| Breast Cancer | ||||
| MDA-MB-231 | 3.4±0.02 | 0.56±0.05 | 50.3±1.7 | 13.6±0.8 |
| SK-BR-3 | 5.4±0.5 | 1.6±0.07 | 25.5±0.24 | 12.3±0.1 |
| BT-474 | 6.6±0.03 | 0.9±0.02 | >1000 | 91.7±8.5 |
| MCF-7 | 3.05±0.6 | 1.02±0.02 | 92±10 | 44.3±2.5 |
| T47D | ND | 0.9±0.16 | ND | 32.3±1.2 |
| Endometrial Cancer | ||||
| An3-CA | ND | 3.8±0.08 | ND | ND |
| Hec-1A | ND | 10.3±1 | ND | ND |
| Prostate Cancer | ||||
| PC-3 ATCC | 3.3±0.8 | 4.9±0.2 | Not toxic | 56.8±0.9 |
| LNCaP | 1.9±0.3 | 1.55±0.08 | 181.5±13.5 | 46.5±2.5 |
| Pancreatic Cancer | ||||
| Panc-1 | ND | 5.4±1.34 | ND | ND |
| Bx-PC-3 | ND | 4.1±0.76 | ND | ND |
| MiaPaCa | 4.07±0.17 | 1.61±0.11 | 44.3±9.9 | 44.4±2.9 |
| Ovarian Cancer | ||||
| OVCAR-3 | 5.8±0.3 | 3±0.5 | 36.45±1.4 | 12.4±1.5 |
| A2780 | ND | 2.02±0.09 | ND | ND |
| SKOV-3 | 50.5±5.3 | 21.1±0.9 | 49.2±3.8 | 28.2±4.9 |
| Kidney Cancer | ||||
| A498 | ND | 2.5±0.5 | ND | ND |
| Non-cancerous | ||||
| 3T3 | >1000 | 121.2±16.5 | >1000 | 102.7±6.8 |
| MCF-10A | ND | 15.3±2.5 | ND | 22.6±2.5 |
Table 2: Efficacy of EP-100 and CLIP71 in various cell lines after 4 and 24 hours. Data are presented as IC50 values in [µM] from Hill Plot analysis. ND=not determined.
Median increase of activity for EP-100 compared to CLIP71 in LHRH receptor positive cell lines was 23 fold and 28.5 fold after 4 and 24 hours. The median difference of activity between cancer and non-cancerous cell lines was 50 fold.
The specificity of EP-100 to cell surface LHRH receptors was further confirmed in a competition experiment in vitro where MDA-MB-435S human melanoma cells were incubated with EP-100 in the presence of increasing concentrations of the LHRH analog, Triptorelin.
Incubation of cells with Triptorelin increased significantly the cell viability (i.e. decreased the cytotoxicity of EP-100 given at 2.5 µM) in a concentration-dependent manner from 13.5±0.6% in the absence of LHRH to 35.8±2.7% (p<0.0002), 48.3±4.2% (p<0.0001), 55.1±7.1% (p<0.0001) and at 100 µM LHRH presence 81.12±3.07% (p<0.0001) (Figure 4). These results suggest that EP-100’s cytotoxicity was mediated by its interaction with the LHRH receptor. Similar results were obtained for pancreatic cancer cell lines in the presence of LHRH analogues (data not shown).

Figure 4: Effect of the LHRH analogue, Triptorelin, on EP-100’s efficacy in the melanoma cell line MDA- MB-435S. EP-100 was added at a concentration of 2.5 µM in the presence of increasing concentrations of Triptorelin at concentrations of 2.5, 10, 50 and 100 µM.
EP-100’s fast acting properties on cancer cells expressing functional LHRH receptors were further evaluated using confocal fluorescence microscopy. One of the mechanisms of action has been described as membrane disintegration through contact of the cationic cytolytic peptide with the negatively charged cancer cell membrane. Another mechanism was described as depolarization of mitochondria through internalized lytic peptides. Two approaches were used to determine the mechanism of action for EP-100 at micromolar concentrations in vitro: 1. EP-100 acting on MDA-MB-435S melanoma cell line visualizing the nucleus, mitochondria and the plasma membrane through fluorescent markers and 2. Fluorescent labeled EP-100 used in a time course experiment in a LHRH receptor positive and a LHRH receptor negative cell line in which the nucleus, mitochondria and EP-100 were labeled with fluorescent markers. The plasma cell membrane of the human melanoma cell line MDA-MB-435S showed significant disintegration after minutes of exposure to 10 µM EP-100 indicating a rapid onset of the cytotoxic action of EP-100 in this LHRH receptor-expressing cell line (Figure 5B-5D). No effects were noted in cells treated with saline (Figure 5A). In a second experiment where 2 µM fluorescent labeled EP-100 was added to SKOV- 3 cells (LHRH R negative, Figure 5E) and MDA-MB-435S cells (LHRH R positive, Figure 5F) the confocal micrographs showed membrane disintegration but also some intracellular uptake in the melanoma cell line as early as 2 minutes. Blebbing of the plasma membrane as early as 30 minutes was observed as well as the formation of vesicles from the outer membrane and fading of mitochondrial dye were observed, suggesting leakage of the cellular complex (Figure 5F). This effect was only minimal in the SKOV-3 cell line with adsorption of EP-100 on the cell surface without any fading of mitochondrial markers (Figure 5E). Lytic peptides consisting of the KLAK sequence have been described as mitochondrial toxins because they depolarized mitochondrial membranes. The effect of EP-100 on mitochondrial depolarization was tested using JC-1 fluorescent indicator. JC-1 is a cationic dye that is widely used to detect mitochondrial membrane potential. The accumulation of JC-1 in polarized mitochondria and emission of red fluorescence (590 nm) is membrane potential dependent. Upon depolarization of mitochondria, JC-1 diffuses into the cell cytoplasm and emits green fluorescence (529 nm).

Figure 5: Mechanism of action of EP-100. Fluorescent confocal micrographs of MDA-MB-435S.luc melanoma cells (green membranes, red mitochondrial and blue nuclear staining). Comparison of membrane intactness in MDA-MB-435S.luc melanoma cells in saline (A) and after exposure to EP-100 after 5 minutes (B), 30 minutes (C) and 60 minutes (D). The cell membrane is rapidly disintegrating in the presence of EP-100 leading to cell death after 60 minutes. E) LHRH receptor negative SKOV-3 cells and F) LHRH receptor positive MDA-MB-435S.luc cells exposed to FITCEP-100. Binding to the LHRH receptor positive MDA-MB-435S.luc cell membrane and disintegration was detectable. Membrane depolarization in JC-1 stained melanoma cells in control (G) and EP-100 (H) exposed MDA-MB-435S.luc cells compared to a mitochondrial depolarization agent, CCCP (I) demonstrates absence of mitochondrial membrane depolarization after EP-100 exposure.
In confocal microscopy images of live untreated control MDA-MB- 435S cells stained with JC-1, bright red stained mitochondria were clearly visible with diffuse green cytoplasmic staining Figure 5G. There was little observable loss in red mitochondrial staining after EP-100 treatment for up to 30 minutes (Figure 5H). CCCP-treated cells lost the distinct red mitochondrial stain and showed a diffuse orange cytoplasmic stain rather than the expected green cytoplasmic stain (Figure 5I).
These observations indicate EP-100 specifically caused membrane disruption in LHRH receptor- positive MDA-MB-435S cells within minutes of exposure without altering the membrane potential of mitochondria. LHRH receptor-negative SKOV-3 cells remained intact following exposure to EP-100.
EP-100 binds with high affinity to the LHRH receptor on the cell surface and kills cells on contact with the cell membrane. LHRH receptors have been found on cell surfaces, the cytosol and the nucleus [59,60]. Immunohistochemistry was used to determine if the target cancer cells were expressing the LHRH receptor on the cell surface. LHRH receptor expression on cancer and non-cancerous cells were determined through immunohistochemistry using a commercially available monoclonal antibody against the LHRH receptor. This assay was used to determine the expression of surface receptors as the functional target for EP-100. Quantification of the surface receptor levels was conducted using a computer assisted imaging program that generated VIAS score numbers for each cell line ranging from 0, 1+, 2+ and 3+. The human ovarian cancer cell line SKOV-3 and the endometrial cancer cell lines KLE and Hec1A tested negative for surface LHRH receptors with VIAS scores of 0. The ovarian cancer cell line OVCAR-3 had a VIAS score of (2+); among breast cancer cell lines T47 D was (1+), MCF-7 (3+), melanoma cell line MDAMB-435S (3+), prostate cancer cell line, PC-3 (2+). Two non-cancerous cell lines represented by MCF-10A breast epithelial (0) and 3T3 mouse fibroblast cells (0) were negative for LHRH receptors.
Correlation of LHRH receptor levels and sensitivities for EP-100 was assessed using literature values for LHRH receptor levels in selected cell lines (Figure 6A) [Leuschner, unpublished data; 46,51,61,62] and receptor levels as determined through IHC as VIAS scores (Figure 6B). Both approaches resulted in linear relationships with R2 values ranging from 0.64-0.82 demonstrating that higher receptor expression levels correlated with a greater sensitivity to EP-100 (Figure 6A and 6B). The sensitivity of the target cells was dependent on the presence of the target receptor.

Figure 6: A) Correlation of EP-100 sensitivities in cancer cells based on Ventana Image Analysis System (VIAS) Scores after immunohistochemistry and B) on LHRH receptor capacities reported from the literature based on Scatchard analyses [46,51,59,61]. Human cancer cell lines, incubated with EP-100 for 24 h, showed variable sensitivities to EP-100, which correlated with LHRH receptor capacities and VIAS Scores with r2 values of 0.64 and 0.82. The LHRH-receptor negative cell line SKOV-3 was used as measure for specificity. SKOV-3 and Hec 1A cells which had no detectable LHRH receptors in immunohistochemistry were insensitive to EP-100 treatment. C) mRNA expression levels in various human cancer cell lines and mouse fibroblast 3T3 cells as negative control. Relative mRNA levels ranged from 10 to 160. D) Correlation of mRNA levels for LHRH receptors from individual cell lines to their sensitivity to EP-100 expressed as micromolar IC50 values. No correlation was detected between LHRH receptor mRNA levels and sensitivity to EP-100 (r2 =10-6).
The LHRH receptor expression based on mRNA levels showed variable expression levels among cell lines including SKOV-3 cells (Figure 6C) but failed to correlate with sensitivities of the cells to EP-100 (Figure 6D) suggesting that target cell identification cannot rely on molecular biological assays-instead surface receptors have to be confirmed in immunohistochemistry methods or direct binding of the ligand.
EP-100 showed maximal activity already after 4 hours of exposure in vitro (Table 2). An activity profile of EP-100 compared to CLIP71 in a LHRH receptor positive ovarian cancer cell line (OVCAR-3) was assessed and compared to a LHRH receptor negative ovarian cancer cell line (SKOV-3). The role of LHRH receptor targeting was also evaluated in this study by comparing the potency of EP-100 to the unconjugated lytic peptide, CLIP71. EP-100 showed maximal potency in destroying cancer cells through a receptor targeted mechanism within less than 1 hour that was far superior to CLIP71.
In OVCAR-3 cells (p34) EP-100 reached its maximal efficacy after 4 hours of incubation (IC50 values [µM] were 6.7±0.01 after 5 minutes; 5.7±0.05 after 15 minutes; 5.3±0.05 after 30 minutes; 1.6±0.06 after 1 hour, 1.5±0.04 after 2 hours, 0.58±0.01 after 4 hours and 0.55±0.001 after 24 hours). In contrast, CLIP71 resulted for the same time points in 40- 50 fold reduced sensitivities with IC50 values [µM] from 337±33.5 after 5 minutes, 126±10.9 (15 minutes), 85.5±15.5 (30 minutes), 71.5±6.0 (1 hour), 52.5±3.5 (2 hours), and 23.1±1.0 after 24 hours (p<0.005). The LHRH receptor negative SKOV-3 cell line had IC50 values [µM] for EP-100 at 95.2±0.8 (15 minutes), 89.8±3.6 (1 hour), 90.8±2.6 (2 hours), 78.1±11.5 (4 hours) and 11.5±1 µM after 24 hours. These sensitivities were similar to CLIP71 for SKOV-3 cells: 93.2±6.3 (15 minutes), 86.7±2.5 (1 hour), 59.8±0.14 (2 hours), 75.4±7.8 (4 hours) and 50.8±0.04 µM after 24 hours. EP-100 was a fast acting agent against LHRH receptor positive cancer cells compared to the unconjugated peptide, CLIP71 (Figure 7A) and was 40- 50 fold more potent in the LHRH receptor positive cells line. The LHRH receptor negative SKOV-3 cell line showed similar kinetic profiles for EP- 100 and CLIP71 (Figure 7B).

Figure 7: Kinetics of EP-100 and CLIP71 in 2 ovarian cancer cell lines in vitro presenting functional LHRH Receptors (A) NIH:OVCAR-3, (B) SKOV-3 ovarian cancer cells that do not express cell surface LHRH receptors. EP-100 exerts its maximal activity within 1 hour whereas CLIP71 shows less potency to kill cells. LHRH receptor negative cells (SKOV-3) did not respond to EP-100 and CLIP71.
These data indicate that EP-100 and CLIP71 kill cells by a different mechanism of action and that receptor targeting significantly increases the efficacy of the lytic peptide (Figure 7A and 7B).
In general, all cell lines expressing functional LHRH receptors such as OVCAR-3 treated with EP-100 showed maximum effect within 0.5-1 hour of incubation whereas 24 hours were required for the maximal effect of CLIP71. Cell lines that did not present cell surface LHRH receptors (SKOV-3 and HEK 1A; Table 2), were resistant to EP-100.
Prior to in vivo efficacy studies, dose finding studies were conducted in nude mice. EP-100 was injected IV as bolus injection at doses of 2, 5, 16 and 25 mg/kg (N=4) and the mice were sacrificed 8 days later. Injections at a dose up to 16 mg/kg of EP-100 were well tolerated with 100% survival. Hematological parameters were unchanged and similar to saline controls (data not shown).
Efficacy of EP-100 in a human ovarian cancer xenograft (OVCAR-3) was assessed with twice weekly dosing on day 33 after tumor cell injection and continued on days 40 and 47 with 0.02 mg/kg (N=10), 0.2 mg/kg (N=10), and 2.0 mg/kg (N=9). Controls received saline injections, and untargeted CLIP71 (2mg/kg, N=10) or cisplatinum (N=10) injected i.p. on days 33, 34 and 35 with a total of 10 mg/kg. A baseline group was sacrificed at treatment start (N=9).
Tumor volumes at treatment begin (day 33) were 189.4±41 mm3 . Tumor weights at treatment begin were 0.152±0.021 g (N=9; baseline group). All treated mice and controls survived the study. Tumor volumes [mm3 ] at necropsy were similar for saline controls 1,147±326.9 mm3 (N=10), for CLIP71 1,682±461 mm3 (N=10) and 1,457±350 mm3 (N=10) for cisplatinum treated mice. Treatment response was measured as tumor volume compared to saline controls, CLIP71 and cisplatinum (Figure 8A and 8B), change of tumor weights compared to baseline values (Figure 8C) and tumor marker CA-125 (Figure 8D).

Figure 8: A and B) Efficacy of EP-100 in drug resistant ovarian cancer xenograft–OVCAR-3. Tumor volumes decreased in xenografted mice treated with 0.02, 0.2 or 2 mg/kg EP-100 once weekly for three weeks. C) Tumor weight changes compared to baseline decreased in all EP-100 treated groups and were below baseline levels after treatments with 0.2 and 2 mg/kg of EP-100. D) Tumor marker CA-125 was significantly reduced in EP-100 treated groups confirming tumor cell death. E and F) PET/CT scans of saline control and EP-100 treated mice show lack of FDG uptake after treatment.
Tumor volumes in EP-100 treated groups decreased in a dose dependent fashion compared to saline controls (1,147±326.9 mm3 (N=10), CLIP71 (1,682±461 mm3 (N=10) and cisplatinum (1,457±350 mm3 (N=10) and were for the lowest dose of 0.02 mg/kg 361.3±196 (p<0.025) significantly lower than saline controls, for 0.2 mg/kg 91.8±22.9 (p<0.0001), for 2 mg/ kg 200.4±36 (p<0.001) at study endpoint. Tumor weights at study endpoint in EP-100 treated groups were decreased in a dose dependent fashion compared to saline controls (0.3373±0.07 g), CLIP71 (0.3867±0.92 g) and cisplatinum (0.3825±0.07 g) and were for the lowest dose of 0.02 mg/kg 0.174±0.052 (p<0.03), 0.2 mg/kg 0.089±0.023 (p<0.003 vs saline controls, p<0.043 vs baseline) and for the 2 mg/kg group 0.1188±0.034 g (p<0.042). Significance was achieved for EP-100 at 0.2 mg/kg when compared to CLIP71 and cisplatinum (p<0.03). Two tumor free mice (2/10 and 2/9) were found in groups treated with 0.2 and 2 mg/kg EP-100.
Tumor weight changes were determined by comparing tumor weights at baseline and study endpoint for each group. Saline controls, CLIP71 and cisplatinum treated mice had tumor weight increases of 0.1873±0.07 g, 0.23±0.09 g and 0.27±0.06 g. Tumor weight reduction at study endpoint in EP-100 treated groups was dose dependent compared to baseline with significant reduction at doses of 0.2 mg/kg -0.06±0.02 g (p<0.012). Tumor weight reduction at doses of 0.02 mg/kg measured 0.024±0.052 g and the 2 mg/kg dose was -0.03±0.03 g reduced compared to baseline values (Figure 8C). Tumor weight changes at doses of 0.2 and 2 mg/kg were not significantly different (p=0.24), whereas the difference between 0.02 and 0.2 mg/kg was significant (p<0.013), indicating that maximal efficacy was reached at 0.2 mg/kg doses.
Tumor growth inhibition (TGI) was greatest at doses of 0.2 mg/kg with TGI values of 71.8, 87.3 and 72.1% at doses of 0.02, 0.2 and 2 mg/kg, respectively. This observation is in agreement with other xenograft models where maximal efficacy was detected in bolus injections of 0.2 mg/kg of EP-100 (unpublished data). In a previous dose response study ranging from 0.0002 to 2 mg/kg tumor volume reduction was observed at doses as low as 0.02 mg/kg. Interestingly, increase of doses greater than 0.2 mg/kg typically did not translate into increased efficacy in the xenograft models tested. At doses up to 1 mg/kg the dose concentration administered were less than 0.25 mg/ml whereas the dose concentration at 2 mg/kg reached 0.5 mg/ml. Dose concentrations greater than 0.5 mg/ml may have affected the integrity of the tail veins at the injection site which may cause insufficient delivery of EP-100 at these higher doses. This is supported by unpublished data obtained in xenograft models, where a dose of 2 mg/ kg was effective when injected at a dose concentration of 0.25 mg/ml by increasing the injection volume.
Viable cells in OVCAR-3 xenografted tumors secreted tumor marker CA-125 and served as measure for response to treatment with EP-100, CLIP71 and controls (saline and cisplatinum groups). Serum CA-125 levels correlated with tumor weights (r2 =0.66). Tumor marker values CA- 125 [U/g tumor weight] were similar for saline controls 2.99±1.13 U/g tumor weight (N=7), 2.7±1.4 U/g tumor weight for CLIP71 treated mice (N=10) and 2.4±0.6 U/g tumor weight in cisplatinum treated mice. In EP- 100 treated groups tumor marker CA-125 values [U/g tumor weight] were significantly reduced with CA-125 levels of 0.8±0.4 (N=10) dose 0.02 mg/ kg (p<0.016), 0.02±0.02 (N=8) for 0.2 mg/kg (p<0.008) and 0.49±0.23 (N=6) at the 2 mg/kg dose (p<0.034) (Figure 8D).
In a second study, OVCAR-3 xenografted mice were treated twice weekly with a total of 6 injections of EP-100 at 0.2 mg/kg doses on days 38, 41, 44, 48, 52, 59 and 61 after tumor cell injection. Tumor weights were decreased in mice treated with EP-100 (0.22±0.03 g) compared to saline controls (0.52±0.9 g) (p<0.0004). Liquid filled cysts were found in 3/16 tumors on day 50 and again in 4/16 tumors on day 59 in the EP-100 treated group.
[18F]-2-fluoro-2-deoxy-D-glucose (FDG) is a contrast agent that accumulates in cells of high metabolic activity and is used for determination and diagnosis of viable tumors in the clinic. Further evidence for treatment responses were observed in PET/CT scans of saline control and EP-100 treated mice with palpable tumors. No [18F]-DG uptake was detected in EP-100 treated animals whereas saline controls showed [18F]-DG uptake (Figure 8E and 8F) confirming that tumors were not viable after EP-100 treatment.
Histological evaluation of tumor sections stained with hematoxylin/ eosin from treated mice showed viable tumor cells in saline controls, mice treated with CLIP71, and cisplatinum. Viable tumor cells with mitotic figures were found in tumors treated with saline or unconjugated CLIP71 (Figure 9A and 9B). Tumor sections of mice treated with EP-100 did not show viable tumor cells. Extensive necrosis and immune cell infiltration were evident in tumors from mice treated with EP-100 at all doses being most prevalent for the 0.2 and 2 mg/kg dose (Figure 9C and 9D).
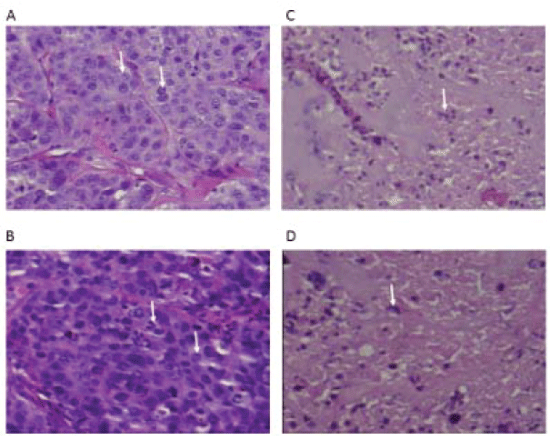
Figure 9: Histology of saline control, CLIP71 and EP-100 treated tumors. Saline controls and CLIP71 groups showed viable tumor cells with many mitotic figures. EP-100 treated tumors were necrotic with macrophage infiltrating cells.
In summary, EP-100 was well tolerated in xenografted mice and resulted in tumor regression, necrosis of tumors administered at 0.2 mg/kg doses in single weekly injections or at 0.2 mg/kg in 6 biweekly injections after 6 weeks. Tumors of EP-100 treated mice were not viable based on tumor marker measurements and PET/CT scan analysis. CLIP71 was ineffective in killing xenografted tumors when administered systemically. EP-100 did not affect CBC or serum chemistry in any of the studies.
EP-100 is a novel, targeted, membrane disrupting peptide that has been selected from over 300 newly designed lytic peptide conjugates based on high anti-cancer activity and low hemolytic activity. EP-100 is a 28 amino acid peptide (MW=3,317 g/mol) comprised of a natural hormone sequence of the luteinizing hormone releasing hormone, joined to an 18 amino acid lytic peptide (called CLIP71). The sequence of EP-100 allows for easy scale up synthesis by standard solid phase chemistry, and up to 500 g quantities have been produced for clinical trials. This paper presents pre-clinical data sets on EP-100 that have been included in the IND submission that have pre-ceded Phase 1 and Phase 2 clinical studies. Part of the data have been presented in poster presentations at international meetings. Part of the data have been generated since the IND submission and are presented in this manuscript.
EP-100 is designed to target and specifically kill cancer cells that overexpress LHRH receptors via a novel mechanism of action that involves direct membrane disruption and necrosis. LHRH and CLIP71 are joined together without a linker to form a cancer cell targeting lytic peptide. Release of the toxic moiety is not required for anti-cancer activity. Because of its wide potential application for cancer treatments, EP-100 has been selected for development towards clinical applications. In this paper, we have demonstrated that 1) EP-100 binds to the LHRH receptor with high affinity, 2) cell killing correlates with surface LHRH receptor expression on the target cancer cells and 3) cell killing occurs within 1-4 hours in LHRH receptor positive cancer cell lines, 4) the mechanism of action involves rapid membrane lysis without depolarization of mitochondria, 5) cancer cells are killed by necrosis, 6) EP-100 is highly potent when administered systemically into ovarian cancer xenograft bearing mice, unlike the unconjugated lytic peptide, and kills xenografted ovarian tumors by necrosis and induces immune cell infiltration.
Increased specificity of cancer therapeutics is desired to improve efficacy and safety of systemically administered drugs. Selective targeting of surface receptors on cancer cells, that are not expressed in peripheral tissues [11,12], ensures specificity and reduces off target effects and damage of vital organs. The LHRH receptor is over-expressed on many solid tumors and hematological malignancies, but is absent on tissues of vital organs, except for the gonads and pituitary gland. It is rapidly internalized and recycled, and demonstrated high efficiency in accumulating compounds through receptor mediated uptake [61,62]. Various LHRH toxin conjugates have been synthesized and showed encouraging pre-clinical activities and biocompatibilities based on specific delivery of the toxins to the target cells. However, these conjugates require highly specific linker technology to ensure stability of the conjugate in circulation and facilitate the release of the toxins within the target cells. This is of particular importance for highly toxic compounds as pokeweed protein [14] and Pseudomonas exotoxin [15], doxorubicin [8], paclitaxel [17], camptothecin [11]. Despite increased specificity, these conjugated toxins caused systemic toxicities and side effects once released into circulation. All these conjugates are in pre-clinical stage except for the LHRH-doxorubicin conjugate, which has been developed for clinical application and is currently being tested in endometrial cancer patients in a Phase III clinical study [10].
Lytic peptide conjugates represent a new class of cancer therapeutics: they act rapidly through membrane disruption and are likely to be independent of drug resistance and independent of cancer cell proliferation. LHRH presents the targeting moiety for the newly designed lytic peptide conjugate, EP-100. Through targeting and binding to the LHRH receptor on the cell surface, EP-100 specifically accumulates on the target cells that facilitated the lytic peptide membrane interaction without cleaving of a linker to release the toxin. This interaction caused membranes to disintegrate, leading to rapid cell death. EP-100’s fast acting mode of action caused cell death within minutes due to rapid, direct interaction with target cell membranes as demonstrated in kinetics of EP- 100 in comparison to unconjugated CLIP71. Generally, in vitro toxicities ranged between 0.5 and 5 micromolar for the conjugated lytic peptide EP- 100 against LHRH receptor positive cancer cell lines, that were obtained within minutes of exposure in vitro. Low micromolar levels of EP-100 have been reached in patients as demonstrated in pharmacokinetic studies in the first human clinical trial at a dose level of 17.6 mg/m2 over 1 hour [56].
The anti-cancer activity of the targeted lytic peptide EP-100 was superior to the unconjugated lytic peptide, as demonstrated in the kinetics of cell killing with maximal effect as early as 4 hours. The half-maximal concentration of EP-100 that killed cancer cells was up to 28 times lower compared to CLIP-71. Moreover, cancer cells that did not express LHRH receptors were equally killed by EP-100 and CLIP71 at higher IC50 values after 24 hours. The potency of EP-100 correlated with LHRH receptor expression levels of cell surface LHRH receptors, but not with mRNA levels of the LHRH receptor. Non-cancerous cell lines were resistant to EP-100 or CLIP71, and both EP-100 and CLIP71 were 50 times less potent compared to cancer cells. These results were similar to findings from Johnstone [63] and Camilio [27]. Most importantly, EP-100 did not cause adverse effects to vital organs when infused into patients twice weekly in repeated infusions over a period of 6 months [56].
The potency of the lytic peptide unit, CLIP71, was comparable to other synthetic, untargeted lytic peptides described in the literature that killed in vitro various cancer cell lines at IC50 values ranging from 2.9- 14 micromolar [27,28,64]. However, a major drawback for unconjugated lytic peptides for clinical development is that they cannot be administered systemically, thus limiting their clinical application to direct injection into the tumors [27,30]. This is in stark contrast to EP-100 and other targeted lytic peptides that can be systemically injected and are suitable for treatment of primary tumors and metastases. EP-100 is suitable for clinical application as it is neither hemolytic nor antigenic, unlike natural lytic peptide melittin or the synthetic peptide LL37. One of the mechanisms of cell killing of EP-100 involved plasma membrane disintegration at low micromolar levels, with no evidence of mitochondrial depolarization. The mechanism of cell killing by lytic peptides can involve the membrane active lysis and, if internalized, the mitochondrial toxicity [65-67]. Gaspar et al. reviewed various sequences and compositions of lytic peptides that showed membrane activity, and or internalization [23]. Both mechanisms were identified as necrosis, which was secondary to plasmamembrane disruption [67]. The lactoferricin analogue, LTX-315, showed membrane disintegration at high concentration of 35 µM and was internalized at low concentrations (3.5 µM) where depolarization of the mitochondrial membrane was observed [68]. Some lytic peptides produced apoptosis, especially if internalization of the peptide occurred [69]. In these cases mitochondrial membrane permeabilization occurred or change of mitochondrial membrane potential [39,70,71]. Two 23 amino acid lytic peptide conjugates targeting LHRH receptors were described by Yates et al., demonstrating cell killing activity of 5-6 and 9-10 µM within 6 hours in prostate cancer cell lines that expressed LHRH receptors. Interestingly these two lytic peptide conjugates exerted different mechanism of action: JCHLHRH (KLA composition) caused membrane rupture and cell bursting; whereas cells exposed to JC21LHRH (KFA composition) showed cell shrinkage without bursting and expression of the apoptotic marker Annexin V [35]. In vivo data were not presented for this lytic peptide conjugate.
Although the detailed mechanism of action for EP-100 has not yet been resolved, the disintegration of cancer cell membranes compared to normal cells may be attributed to their surface membrane characteristics. The positive charge of lytic peptides builds the basis for their high potency in destroying negatively charged membranes, such as cancer cell membranes.
Surface membranes of eukaryotic cells have an asymmetrical distribution of lipids which determines their surface charge [72,73]. Cancer cells are negatively charged as they contain up to 8 fold higher levels of the negatively charged membrane lipid phosphatidylserine (PS) in their outer membrane, whereas normal cells have a neutral outer membrane and contain PS only in the inner plasma membrane [73-76]. This distribution of charge resulted in greater sensitivity of a cancer cell membrane to a cationic cytolytic peptide. Normal cells have a neutral cell membrane that does not promote interaction with the cationic peptide moiety, their membrane cholesterol prevents peptide insertion, leading in early proteolytic destruction of the cytolytic peptide [77,78]. The presence of heparan sulfate on the cancer cell surface may have an effect on the interaction of the lytic peptide with the membrane due to neutralization of the negative charge [79]. No data have yet been generated to determine if that is the case for EP-100 and similar lytic peptide conjugates.
Rapid cytotoxicity is generated through direct interaction of the lytic peptide with the cell membrane (for review see [20,21]. This is achieved by conjugating a ligand to the cytolytic peptide to increase specificity to the target cells and facilitate concentration and accumulation on the cell surface membrane through receptor-ligand binding necessary to destroy the cell membrane. In summary, the rapid cell killing properties and specificity of EP-100 to kill cancer cells rather than normal cells is facilitated through expression of the LHRH receptor on the cancer cell surface, along with the high negative charge of the cancer cell membranes through PS accumulation. Furthermore, the cholesterol content in normal cells protects from lytic peptide attacks, even in normal cells that express LHRH receptors like the pituitary and gonads. The pituitary cells that express LHRH receptors release LH and FSH. Pharmacodynamic effects of EP-100 on the pituitary has been observed and caused reduction of release of LH and FSH during EP-100 treatment of patients. LH and FSH production was re-instated upon discontinuation of EP-100 treatment [56].
In in vivo studies, EP-100 was highly active when administered systemically. EP-100 demonstrated effective and specific cell killing and caused reduction of tumor volumes and tumor weights and reduction of live tumor cells with evidence of necrosis detected in tumor sections of treatment groups. EP-100 treated tumors had significant immune cell infiltration. In contrast, unconjugated lytic peptide CLIP71 or LHRH peptide was not active in vivo when systemically injected. CLIP71 had no effect on tumor histology or tumor burden. EP-100 demonstrated superior activities in human ovarian cancer xenografts in vivo when administered once weekly for 3 weeks (at doses as low as 0.02 mg/kg). Six injections over a time course of 4 weeks at a dose of 0.2 mg/kg of EP-100 resulted in tumor free mice and decrease in tumor viability as confirmed by histology, PET/CT and reduction of tumor marker CA- 125. Compared to the first generation of targeted lytic peptide conjugates, Hecate-LHRH and Phor21-LHRH, EP-100 was superior: EP-100 was effective at lower doses in xenograft models (0.02 mg/kg compared to 12 mg/kg in a prostate cancer xenograft model and 10 mg/kg in a pancreatic cancer xenograft model) [41-49,77]. The first generation lytic peptide conjugates, Hecate-LHRH (<10 µM) and Phor21-LHRH (21 µM) were more hemolytic compared to EP-100 (>300 µM) and were less suitable for clinical development [unpublished data from the author]. EP-100 was well tolerated in mice even after repeated injections at doses up to 400 fold higher (8 mg/kg) than the therapeutically effective doses in ovarian cancer xenografts (0.02 mg/kg). No toxicities to liver or kidney were observed in treated mice as determined in gross pathology and serum chemistry, body weights remained stable in treated mice indicating that EP-100 showed high specificity in xenograft models. The effective dose for ovarian cancer xenografted nude mice was as low as 0.02 mg/kg that was 10 fold better compared to the Phor21 containing conjugate [33]. The dose limiting toxicity was reached at doses of 25 mg/kg of EP-100 in nude mice and was 8 mg/kg in repeat dose studies in CD1 mice. The therapeutic window was at least 40-fold for EP-100 systemic administration based on toxicology data conducted prior to Phase 1 clinical trial in humans.
Other conjugates of CLIP71 showed similar properties to EP-100. The conjugation of CLIP-71 to FSH for targeting FSH receptors on prostate cancer xenografts showed that low doses of 0.2 mg/kg were effective in killing cancer cells by necrosis, and in addition reduced neovasculature expressing the target receptor [80].
Several lytic peptide conjugates are at pre-clinical stage: A fourteen amino acid lytic peptide of the D amino acid containing sequence, (KLAKLAK)2 , has been conjugated to ligands such as PSMA [36], anti-CD33, anti-CD 19 [37], tumor and neovascular targeting peptide RGD [38], Her2-targeting peptides [40] and a gastrin targeting peptide conjugate [39] that have demonstrated specificity in the low micromolar range in vitro and shown tumor growth arrest after intravenous injections of doses of 3 mg/kg in xenograft models. A synthetic lytic peptide (D-K6 L9 ) conjugated to a EGFR targeting peptide showed specificity in killing various human cancer cell lines at concentrations between 6.5- 12 µM compared to the untargeted lytic peptide 16-32 µM [34]. Cell killing was rapid within 1 hour. Assessment on the mechanism of action showed that EGF-D-K6 L9 killed target cells through apoptosis. Systemic intravenous injection of EGF-D-K6 L9 killed xenografted breast and pancreatic cancer cells within 3 weeks at doses of 2, 5 and 10 mg/kg [34]. Compared to the above summarized examples, EP-100 presents a much more potent compound killing cancer cells in vitro at low micromolar levels. The composition of the lytic moiety being phenylalanine generated necrosis rather than apoptosis as seen for various lytic peptides containing leucine [34-36].
In summary, EP-100, CLIP71 conjugated to LHRH without a linker, is a highly selective, potent peptide conjugate. EP-100 binds to the LHRH receptor with high affinity, has a wide therapeutic margin, fast acting molecule that is not antigenic and kills rapidly LHRH receptor expressing cancer cells at low micromolar levels that are achievable in humans. EP- 100’s rapid action is based on disintegration of the cell membrane. EP-100 does not contain any linkers and exerts its cell killing potency without the need for releasing the lytic peptide domain unlike small molecules conjugated to LHRH that require internalization and intracellular release of the toxins through linker cleavage. In vivo studies demonstrated cell killing through necrosis, resulting in tumor volume and weight decrease as well as immune cell infiltration. Due to the favorable safety profile of EP- 100 and its high potency to kill LHRH receptor positive cancers EP-100 may have potential in clinical applications for ovarian cancers including multi-drug resistant cancers. Safety data have been obtained in two clinical trials with EP-100 in 76 patients for up to 18 months, demonstrating lack of immunogenicity, absence of organ toxicities and bone marrow suppression as single agent (NCT0094955) or in combination with Paclitaxel in advanced ovarian cancer patients (NCT01485848).
The decision to choose ovarian cancer as the first clinical indication was made based on the highly unmet need that exists for advanced, multidrug resistant ovarian cancer patients whose tumors failed to respond to chemotherapy with a clear path to approval. With LHRH receptor expressions of up to 80% in ovarian cancers EP-100 presents a valid candidate for this patient group.
EP-100’s specificity to kill LHRH receptor positive cancers using a novel mechanism of action offers the opportunity to expand the clinical indications to breast, prostate, pancreatic, endometrial cancers, as well as to melanoma and hematological malignancies.
The authors thank Dr. Deborah Vollmer Dahlke for helping preparing the manuscript. We thank Dr. Steve Bujenovic, Director of nuclear medicine OLOL, PET, SPECT and CT Imaging, Baton Rouge, LA and Dr. Kenneth (Kip) Matthews II, PhD DABR, Louisiana State University, Department of Physics and Astronomy Baton Rouge, LA for facilitating the PET imaging and analysis.
Download Provisional pdf here
All Sci Forschen Journals are Open Access