
Table 1: XPO1 inhibition induced cytotoxicity in NSCLC cell lines independent of genotype
Marsha Crochiere* William Senapedis Trinayan Kashyap Tami Rashal Dilara McCauley Michael Kauffman Sharon Shacham Yosef Landesman
Karyopharm Therapeutics, Inc., Newton, Massachusetts, USA*Corresponding author: Marsha Crochiere, Karyopharm Therapeutics, Inc., 85 Wells Avenue, Newton, MA, 02459, USA, Tel: 617-658-0544; E-mail: mcrochiere@karyopharm.com
Article Type: Research Article
Citation: Crochiere M, Senapedis W, Kashyap T, Rashal T, McCauley D, et al. (2016) The Selective Inhibitor of Nuclear Export Compound, Selinexor, Inhibits NF-κB and Induces Anti-Non-Small Cell Lung Cancer Activity Regardless of p53 Status. Int J Cancer Res Mol Mech 2(2): doi http://dx.doi. org/10.16966/2381-3318.126
Copyright: © 2016 Crochiere M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Non-small cell lung cancer (NSCLC) has poor prognosis even with various treatment options. Unfortunately, resistance develops quickly and novel therapies are needed. Exportin-1 (XPO1) is a nuclear export protein upregulated in many types of cancers. Inhibition of XPO1 by selinexor (KPT-330), an oral Selective Inhibitor of Nuclear Export (SINE) compound, leads to nuclear accumulation of tumor suppressor proteins (TSPs), cell cycle arrest and cancer cell death. Selinexor is being evaluated in Phase I and II clinical trials in many different cancer indications (see clinicaltrials.gov for details) including lung cancer (NCT02351505). To date, selinexor has been given to >1400 patients and has shown good tolerability with manageable side effects. In this study the effects of selinexor on NSCLC cell growth and apoptosis were evaluated in vitro and in vivo. Selinexor inhibited tumor cell growth and clonogenic formation and induced cell cycle arrest and apoptosis in cell lines regardless of genetic background (EC50: 25-700 nM). The XPO1 cargo proteins p53, p21, IκB, E2F4, and survivin demonstrated strong nuclear localization after 4 to 24 hour treatment with selinexor followed by apoptosis (i.e. PARP, caspases-3 and -8 cleavage). Selinexor altered the localization of the NF-κB inhibitor IκB and functional evaluation of NF-κB in A549 and NCI-H1299 revealed transcriptional repression with EC50 values similar to the inhibition measured in cell proliferation assays. A549 xenografts in mice treated with selinexor showed markedly greater tumor growth inhibition (%TGI=81% at 20 mg/kg selinexor vs vehicle, p<0.0001) versus cisplatin-treated mice (%TGI=13% at 5 mg/kg cisplatin vs vehicle, p=0.06) when compared to vehicle. Using IHC on paraffin embedded tissue, treated tumors showed reduced cell proliferation (Ki67) while inducing nuclear accumulation of p53, p21, FOXO1, survivin, NF-κB and IκB. In NSCLC, selinexor forces nuclear retention of TSPs, inhibits tumor growth and NF-κB transcriptional activity, and induces cell death regardless of p53 status. These data demonstrate therapeutic potential for the treatment of NSCLC.
XPO1; NSCLC; Selinexor; TSPs; Export; Cancer
Lung cancer is the leading cause of cancer related mortality in the United States. Non-small cell lung cancer (NSCLC), which includes adenocarcinomas, squamous cell, and large cell carcinomas, encompass ~85% of all lung cancers [1,2]. Currently, available regimens such as surgery, chemotherapy, radiation, and targeted therapies provide limited benefit with the 5-year overall survival rate of ≤ 10% in patients with advanced disease and >70% in patients with stage 1 disease. The design of novel, effective and well-tolerated agents is confounded by the complex, poorly understood molecular mechanisms involved in the genesis of the various types of NSCLC. In addition, most lung cancers present as disseminated disease and harbor multiple mutations, rendering them initially or rapidly resistant to available therapies. In NSCLC, genomic studies of tumor tissue suggest that p53 status may be a prognostic indicator of treatment outcomes. Because the p53 pathway is involved in initiating cell cycle arrest and apoptosis upon DNA damage by chemo- or radiation therapy, NSCLC patients harboring mutations (42%) or polymorphisms (42%) in p53 or overexpression of the p53 inhibitor MDM2 may reduce the benefit of standard treatments [3]. Therefore, therapies that are not dependent on functional p53 are highly warranted for NSCLC.
Among the major requirements for lung cancer cell transformation are inactivation of tumor suppressor proteins (TSPs) such as p53 [4] and aberrant activation of cell survival pathways such as NF-κB signaling [5]. Disruption of normal TSP and NF-κB signaling can be achieved through inactivating mutations and/or changes in subcellular localization of proteins in these pathways (i.e. p53, FOXO, IκB, etc.) [6-8]. Transport of proteins across the nuclear pore complex is carried out by a family of proteins called karyopherins; importins transport cargoes containing a nuclear localization signal into the nucleus while exportins transport cargo proteins (as well as specific RNA molecules through RNA binding proteins) with a nuclear export signal from the nucleus to the cytoplasm [9-11]. Most TSPs regulate transcription and require nuclear localization in order to carry out their tumor suppressing functions. Additionally nuclear retention of IκB serves to inhibit the transcriptional activity of NF-κB [7]. Therefore, the nuclear export of TSPs and IκB is an efficient way to neutralize their function and promote tumor cell growth.
Although there are seven known nuclear export proteins, exportin 1 (XPO1; Chromosome Region Maintenance 1; CRM1), is thought to be the nuclear transporter of most TSPs such as p53 [12,13], FOXO [14], and pRB [15], as well as >200 other cargos [16]. Overexpression of XPO1 protein has been observed in a variety of cancers, including lung cancer, and correlates with poor prognosis [17,18]. Inhibition of XPO1 by leptomycin B (LMB), a highly potent, natural product inhibitor of XPO1, inhibited lung cancer cell growth and induced cytotoxicity in both p53 wild type (wt) and p53 mutant tumors, with less effect on normal bronchial epithelial cells [13]. Although these data suggested the potential usefulness of targeting XPO1 for lung cancers, LMB (elactocin) was shown to be ineffective in the clinic due to marked toxicities in humans that were not thought to be related to XPO1 inhibition [19,20]. More recently, inhibition of XPO1 by the potent, small molecule, Selective Inhibitor of Nuclear Export (SINE) compound selinexor (KPT-330), has shown broad preclinical anticancer activity and specificity [21] and is currently being evaluated in Phase I and II clinical trials in multiple different cancer indications (see clinicaltrials.gov) including lung cancer (NCT023515050). Additional SINE compounds KPT-185 and KPT-276, were reported to induce selective anticancer cytotoxicity against non-small cell lung cancer (NSCLC) in vitro and in vivo [22]. However, the mechanistic and in vivo effects of selinexor on NSCLC are not yet well established. Here, we present the effect of selinexor on NSCLC and demonstrate that XPO1 is a potential target for the treatment of this malignancy.
Cell lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 100units/mL penicillin, 100 μg/mL streptomycin (Gibco), and 1X GlutaMAX (Gibco), and maintained in a humidified incubator at 37°C in 5% CO2 . NCI-H226, NCI-H520, NCI-H889, NCI-H1299, A549, NCI-H2122, and NCI-H2030 were obtained from ATCC (Manassas, VA). Selinexor was synthesized by Karyopharm Therapeutics, Inc. (Newton, MA).
~10,000 cells from log phase cultures were seeded in 100 μl per well of 96-well flat-bottom culture plates. Escalating concentrations of selinexor were added to the wells and incubated at 37°C in a humidified incubator with 5% CO2 for 72hours (in triplicate). Then, 100µl of MTT (3-4, 5-dimethylthiazoyl)-2, 5-diphenyltetrazolium bromide 5mg/mL, Promega) was added to each well and incubation was continued for 4hours at 37°C. In each well, 100µl/well of solubilization solution, containing DMSO and Sorenson buffer, were added. After complete solubilization of the dye, plates were read at 570nm on an ELISA reader. The mean optical density (OD) ± SD for each group of replicates was calculated. The whole procedure was repeated three times. The inhibitory rate of cell growth was calculated using the formula: % Growth inhibition=(1-OD extract treated)/OD negative control × 100 [23].
NCI-H1299 and A549 cells were plated at 5000 cells/well in 12 well plates (Cell Treat). The following day cells were treated with DMSO (Sigma), 1 µM selinexor for NCI-H1299 or 5 µM selinexor for A549. On days 0, 4, 6, and 8 cells were fixed and stained with Gentian Violet (RICCA Chemical Company) and imaged with a digital camera (Sony Cybershot). Cells were then solubilized in 10% acetic acid (Fisher Scientific) and the OD was measured at 590 nm.
Cell cycle profile analysis was performed using the BrdU Flow Kit (BD Pharmingen) according to the manufacture’s protocol. Briefly, NCI-H1299, A549, and NCI-H2030 cells were plated in 6 well plates at 500,000 cells/well for day 1 and 200,000 cells/well for day 3. Cells were treated with either DMSO or 1 µM selinexor for NCI-H1299, 5 µM selinexor for A549, and 10 µM and 60 µM selinexor for NCI-H2030. Prior to harvesting, NCI-H1299 and A549 cells were incubated with 10 µM BrdU for 2 hours while NCI-H2030 cells were incubated with 10 µM BrdU for 4 hours. Cells were fixed and stained for BrdU and 7-AAD according to the manufacturer’s protocol. Cells were then analyzed on a BD LSRFortessa (BD Biosciences) at the Dana Farber Cancer Institute (Boston, MA) and the data was subsequently analyzed using FCS Express 4 software (De Novo Software).
NCI-H1299 and A549 cells were plated at 375,000 cells/well in 6 well plates and treated with either DMSO (0) or 30, 100, 300, 1000 or 3000 nM selinexor for 24 and 48 hours prior to collection by trypsinization. Proteins were extracted from cells in Pierce RIPA buffer (Thermo Scientific) supplemented with phosphatase and protease inhibitors (Roche). Protein amounts were determined by the Pierce BCA Protein Assay Kit (Thermo Scientific) and samples were normalized such that for each sample 10 μg of total protein was loaded per lane. Proteins were separated by loading on Novex NuPAGE 4-12% Bis-Tris Gels (Life Technologies) and transferring to nitrocellulose with the Novex iBlot Gel Transfer Stacks (Life Technologies). The following primary antibodies were used for immunoblot analysis: PARP (Cell Signaling), β-actin (Santa Cruz), caspase 3 (Abcam), caspase 8 (Cell Signaling), caspase 9 (Cell Signaling), XPO-1 (Santa Cruz), p53 (Santa Cruz), p21 (Abcam), E2F4 (Santa Cruz), FOXO3a (Cell Signaling), survivin (Abcam), NF-κB (Cell Signaling), and IκB (Abcam). Protein signals were detected with infrared linked species-specific secondary antibodies (LI-COR Biosciences). Western blot images were detected with the ODYSSEY Infrared Imaging System (LI-COR Biosciences).
NCI-H1299 and A549 cells were placed on glass coverslips (BioCoat, BD Biosciences) at 375,000 cells/well in 6 well plates and grown overnight. Cells were treated with 1 µM selinexor for either 4 hours to detect p53 and IκB or for 24 hours to detect p21, E2F4, and survivin. After treatment, coverslips were washed with 1X PBS (phosphate buffered saline) then fixed in either 3% paraformaldehyde buffer (3% paraformaldehyde/2% sucrose/1X PBS) or 100% ice-cold methanol for 15 minutes then washed with 1X PBS. Cells were permeabilized with 0.1% Triton X-100/1% BSA/1X PBS (PFA fixation) or 0.1% Tween 20/0.3 M glycine/1% BSA/1X PBS (Methanol fixation) for at least 30 minutes. After washing 3 times with 1X PBS, cells were stained overnight with corresponding antibodies list above diluted in 1%BSA/1X PBS. Protein signal was detected with species specific Alexa Fluor 488 secondary antibodies (Invitrogen) while DNA was stained with DAPI (Invitrogen). Protein localization was visualized with a Nikon Eclipse Ti inverted fluorescence microscope (Nikon) and monochrome camera (ANDOR).
SCID mice were purchased from National Cancer Institute of Singapore (Dr. Shin Leng). All animal experiments were carried out in accordance with the guidelines of the guide for animal welfare and with the approval of the IACUC. Sixty (60) female CB-17 SCID mice (Charles River Labs strain code 236), aged 5 to 6 weeks were used. The mean body weight prior to treatment was 16.3 grams. The mice were inoculated subcutaneously in the left flank with 4 x 107 A549 cells. A549 (ATCC#CCL-185) NSCLC cells were obtained from ATCC. Treatment was initiated when the tumors reached a mean volume of 98.5 mm3 (SD=21.2 mm3 ). Mice were allocated to six (6) groups of ten (10) mice such that mean tumor volume in each group was within the range of 95 to 104 mm3 . Mice were treated with vehicle, standard of care drugs/positive control drug (cisplatin) or selinexor (KPT-330) at 10 or 20 mg/kg. Vehicle and selinexor were given on a Monday, Wednesday and Friday schedule beginning on Day 1, cisplatin was given by IP injection on Days 1 and 15. Animals’ weights and condition were recorded daily, and tumors were measured on Monday, Wednesday and Friday. Analysis of the tumor volume data was conducted by measuring the Mean Area Under the curve (AUC) for each tumor and comparing the groups using a one-way ANOVA test.
Tumors from the xenograft study were fixed and embedded in paraffin. Sections were cut and routine immunohistochemistry (IHC) was performed. Briefly, sections were deparaffinized through a xylene and alcohol series then treated with 3% hydrogen peroxide. Antigens were retrieved using the Cell Marque detection system (Cell Marque). Antigens were detected by loading slides onto Biogenex I6000 automated immunostainer (Biogenex Laboratories) programmed for each appropriate antibody. Briefly, the program included a 20 minute background blocker, a PBS rinse, a 15 minute Biogenex Avidin block, another PBS rinse, followed by a 15 minute Biogenex Biotin block, a PBS rinse, a 20 minute Biogenex Power Block, then the slides were blown clear of blocker. Primary antibody was applied and incubated for 1 hour at room temperature and then rinsed with PBS. The Cell Marque Detection System was then used to apply Biotinylated Link and was incubated for 20 minutes followed by a PBS rinse. Next streptavidin label was applied and incubated for 20 minutes followed by a PBS rinse. Finally, the Cell Marque AEC chromagen was applied and incubated for 5 minutes and then rinsed twice with deionized water and once with PBS. Slides were transferred to tap water, counterstained in Hematoxylin for 10 dips, and then rinsed with tap water, followed by application of bluing reagent for 10 dips and rinsed with tap water. Cell Marque aqueous mounting media was added and slides were allowed to dry at room temperature overnight or for 20 minutes in an oven. Coverslips were mounted after a quick dip in xylene once dry. Antibodies used for detection of proteins by IHC were the following: Ki67 (Cell Marque), p53 (Santa Cruz), p21 (Cell Signaling), FOXO1 (Cell Signaling), survivin (Abcam), NF-κB (Santa Cruz), and IκB (Abcam).
NCI-H1299 and A549 cells were plated at 200,000 cells/well in 12- well plate and cultured as described above. Cells were pre-treated with serial diluted (starting at 30 µM; 1:3 dilution) selinexor for 1 hour and then exposed to 20 ng/ml TNFα (Peprotech) for 4 hours in serum free media. After the treatment, the cells were washed with PBS and lysed with RIPA buffer. The transcription activity of NF-κB in the cells was measured with the Chemiluminescent Transcription Factor Assay kit (Thermo Scientific) according to the manufacturer’s instruction. Briefly, 1.5 mg/ml of RIPA lysed whole cell extract from each treatment was incubated in a 96 well plate bound with NF-κB biotinylated-consensus sequence. The active NF-κB transcription factor bound to the consensus sequence was incubated with NF-κB p65 primary antibody and then with a secondary HRP-conjugated antibody. A chemiluminescent substrate was added to the wells and the resulting signal was detected using a luminometer. XLFit model 205 was used to calculate IC50 curves.
In phase 1 clinical studies of patients with advanced solid tumors dosed from 3-85 mg/m2 (2-50 mg), the Cmax of selinexor in the serum ranged from 30-1373 ng/mL [24]. To test whether selinexor inhibited the growth of lung cancer cells at clinically relevant concentrations, seven NSCLC cell lines were selected and their ability to proliferate and susceptibility to death were evaluated. Of the 7 cell lines tested in a modified MTT cytotoxicity assay, 5 were sensitive (NCI-H2122, NCI-H226, NCI-H520, NCI-H889, NCI-H1299; EC50<400 nM), 1 had intermediate sensitivity (A549; EC50 400-1000 nM), and 1 was resistant (NCI-H2030; EC50 9100 nM) (Table 1). In addition, sensitivity to selinexor was not associated with the NSCLC subtype nor with the mutational status of p53 or KRAS. All cells were wild type for EGFR and for AKT (Table 1). These results were consistent with findings in other NSCLC cells where sensitivity to selinexor did not correlate with common genetic abnormalities [25].
Table 1: XPO1 inhibition induced cytotoxicity in NSCLC cell lines independent of genotype
To further examine the effect of selinexor on proliferation and cell viability, one sensitive NSCLC cell line, p53 null NCI-H1299 (EC50: 130 nM), and one intermediately sensitive NSCLC cell line, p53 wild type A549 (EC50: 410-700 nM), both of which carry KRAS mutations, were chosen for subsequent analyses. According to the NCI-Navy Medical Oncology Branch, NCI-H1299 are characterized as NSCLC epithelial cells morphologically derived from the lymph node [26] whereas A549 are an adenocarcinoma alveolar basal epithelial cell line [27].
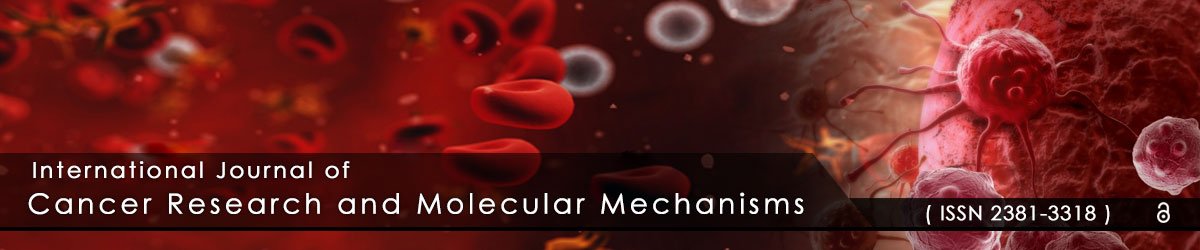
To evaluate the inhibitory effects of selinexor on the growth of tumor cells in culture, the clonogenic assay [28] was used. The effects of selinexor on NSCLC clonal and cell growth were tested over the course of 8 days in culture. Complete inhibition of cell growth by selinexor was demonstrated in both the sensitive NCI-H1299 and intermediately sensitive A549 cell lines (Figure 1). Treatment of NCI-H1299 with 1 µM selinexor (Figure 1A) and A549 with 5 µM selinexor (Figure 1B) for 8 days resulted in complete growth inhibition as determined by visual inspection as well as by spectrometer quantification of Gentian Violet staining of adherent viable cells. In contrast, the vehicle control treated NCI-H1299 and A549 cells grew normally until day 6 when they became over confluent and began to decrease due overcrowding and death (Figure 1). Lower concentrations of selinexor were also tested on both cell lines in the clonogenic assay yielding similar results (data not shown).
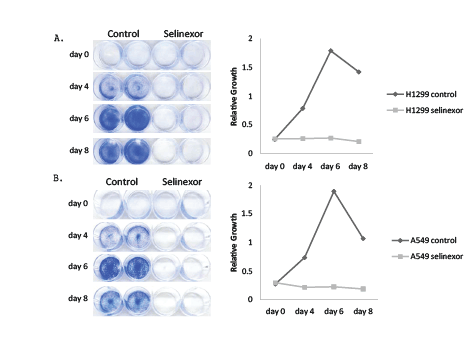
Figure 1: Selinexor inhibited cell growth in NSCLC cell lines.
A) NCI-H1299 and B) A549 cells were plated at 5000 cells/well and treated with either DMSO (control) or with 1 µM selinexor (NCI-H1299) and 5
µM selinexor (A549). Cells were stained with Gentian violet and imaged at days 0, 4, 6, and 8. The control cells for both NCI-H1299 and A549 grew
normally until day 6 when they became overpopulated. The relative growth for both cell types was determined by solubilizing the cells with 10% acetic
acid and measuring the OD at 590 nm. Lack of color detection indicated that treatment with selinexor inhibited cell growth in both NCI-H1299 and
A549 cells.
Growth inhibition by selinexor could potentially be achieved by various mechanisms. In order to better understand these mechanisms, both vehicle and selinexor treated NCI-H1299 and A549 cells were subjected to cell cycle analyses. For these analyses, NCI-H1299 and A549 were treated with 1 and 5 µM selinexor, respectively, for 1 and 3 days and were evaluated for BrdU incorporation and DNA content by flow cytometry (Figure 2). Applying selinexor to sensitive p53 null NCI-H1299 cells reduced but did not eliminate S phase, induced G1 arrest, and increased the sub-G1 population in a time-dependent manner (Figure 2A). In contrast, treating p53 wild type A549 cells with selinexor completely eliminated S phase, induced prominent G2 arrest as well as G1 arrest, and increased sub-G1 (apoptotic) cells, although not to the same extent as observed in selinexor-treated NCI-H1299 cells (Figure 2B). Increase in the sub-G1 cell population correlates with increased cell death, indicating that NCI-H1299 cells had a higher degree of cell death than A549 cells. These data raise the possibility that p53 was involved in the prominent G2 arrest observed in the A549 cells.
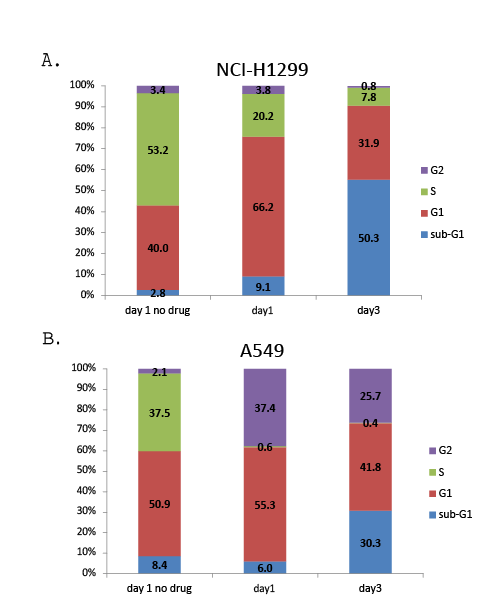
Figure 2: Selinexor induced growth arrest and cell death in NSCLC cell lines. A) NCI-H1299 and B) A549 cells were treated with control (no drug) or with 1 µM selinexor (NCI-H1299) or 5 µM selinexor (A549) and incubated with BrdU for 2 hours prior to collection and fixation at days 1 and 3 in culture. Fixed cells were stained for both BrdU and 7-AAD incorporation and then the cell cycle profile for each cell type was evaluated by flow cytometry.
Additional flow cytometry was also performed on the most resistant NSCLC cell line, NCI-H2030 (EC50 9.1 μM) (Supplementary Figure 1). Cell cycle profile analysis of the resistant NCI-H2030 cell line treated with 10 µM of selinexor for 3 days revealed a reduction in S phase with a corresponding modest increase in G1 phase without significant cell death (Supplementary Figure 1A). Although not clinically relevant, treatment of NCI-H2030 cells with 60 µM selinexor for 2 days reduced S and G1 phases, and increased G2 phase and cell death (sub-G1 phase) (Supplementary Figure 1B). By day 3, 60 µM selinexor caused death in all NCI-H2030 cells (data not shown). These results demonstrated that selinexor administered at a higher dose induced cell cycle arrest and cell death in a time and dose dependent manner similarly in the resistant NCI-H2030 cells as in the sensitive NCI-H1299 and intermediately sensitive A549 cells.
The results from the clonogenic assay and flow cytometry analyses indicate that selinexor induced NSCLC cell death. To determine whether selinexor induced NSCLC apoptosis, apoptotic markers were analyzed by Western blot. NCI-H1299 and A549 cells were treated with 0, 30, 100, 300, 1000 or 3000 nM selinexor for 24 or 48 hours (Figure 3). In NCI-H1299 cells, treatment with >300 nM selinexor for 24 hours led to a reduction of full length caspase 3 and PARP as well as to an increase in their cleaved products. This pattern was also seen in the A549 cells, but higher selinexor concentrations were required for caspase 3 and PARP cleavage consistent with results in other assays. Interestingly, distinctions were observed between intrinsic and extrinsic apoptotic pathways, where the cleavage products of caspase 8 (extrinsic) were more prominent in A549 than in NCI-H1299 cells, and the levels of full length caspase 9 (intrinsic) were reduced in both cell types with selinexor treatment (Figure 3). These results suggest that selinexor induced both the extrinsic and intrinsic apoptotic pathways in NSCLC cells, ultimately resulting in cell death. In addition, induction of apoptosis by selinexor is independent of p53, which is consistent with the effect of SINE compounds on apoptosis in solid tumor and lymphoma models [29,30].
Figure 3: Selinexor induced cell death via apoptosis. Immunoblots of whole cell lysates from A549 and NCI-H1299 cells treated with 0, 30, 100, 300, 1000, or 3000 nMselinexor for 24 (PARP and corresponding actin) and 48 hours (caspases 3, 8, 9 and actin). CL =cleaved, FL =full length.
Immunofluorescence microscopy was used to elucidate the role of selinexor on the nuclear accumulation of key TSPs and cell cycle regulators. In contrast to predominantly cytoplasmic localization in vehicle treated cells, nuclear localization of IκB, E2F4, and survivin was enhanced in NCI-H1299 cells treated with 1 µM selinexor (Figure 4A). Because NCI-H1299 is a p53 null cell line (Table 1), p53 and p21 (under both p53 -dependent and -independent transcriptional control) could not be detected with or without selinexor treatment. A549, a p53 wild type cell line, exhibited substantially enhanced nuclear localization of tested TSPs – p53, p21, IκB, E2F4 and survivin– upon treatment with 1 µM selinexor (Figure 4B). These results demonstrated that selinexor induced nuclear localization of several TSPs regardless of p53 status and induced apoptosis potentially through blocking cell cycle regulators such as E2F4 and survivin in the nucleus.
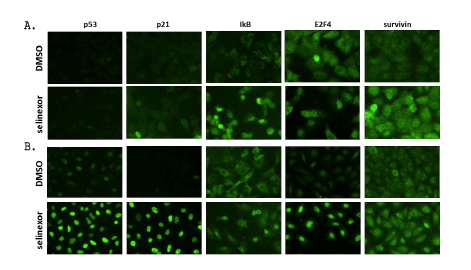
Figure 4: Selinexor induced nuclear localization of multiple TSPs and transcriptional regulators in two NSCLC cell lines, A) NCI-H1299 and B) A549. NCI-H1299 and A549 were treated with 1 µM selinexor for 4 hours (p53 and IĸB) or 24 hours (p21, E2F4, and survivin) prior to fixation and subsequent immunofluorescence microscopy.
Western blot analysis was used to evaluate the effects of selinexor on levels of TSPs and other cell cycle regulator proteins. NCI-H1299 and A549 cells were treated with increasing amounts of selinexor for 24 hours and the protein levels of TSPs and other transcriptional modulators were analyzed. Although the observed dose-dependent decrease of XPO1 in both A549 and NCI-H1299 in response to treatment with selinexor was expected and consistent with previous published results [22], the dose-dependent decrease of FOXO3 a protein was unexpected and not previously observed in other cell lines (Figure 5). In A549 cells, levels of p53 and p21 protein increased in a dose-dependent manner whereas similar to Figure 4A neither protein was detected in the NCI-H1299 cells (Figure 5). E2F4 protein levels in A549 cells were not affected by selinexor treatment whereas in NCI-H1299 cells it was slightly decreased when treated with higher selinexor concentrations. Levels of survivin protein were not altered in either cell type with selinexor treatment (Figure 5). Together these data demonstrate that selinexor can modulate both protein localization as well as expression in order to inhibit cell cycle progression and induce apoptosis.
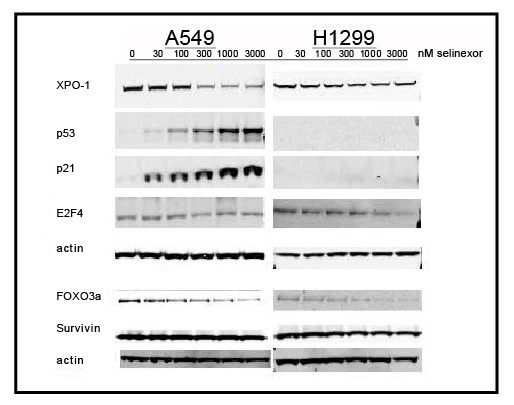
Figure 5: Selinexor inducedprotein expression changes in several TSP and transcriptional regulators in two NSCLC cell lines. Immunoblots of whole cell lysates from A549 and NCI-H1299 cells treated with 0, 30, 100, 300, 1000, or 3000 nMselinexor for 24 hours.
The NF-κB/IκB pathway has been extensively studied for its role in cancer manifestation and progression [31]. Based on the results showing that selinexor treatment induces nuclear accumulation of IκB in vitro (Figure 4), the effects of selinexor on NF-κB activity and protein expression levels in NSCLC cells in vitro was evaluated (Figure 6). NCI-H1299 and A549 cells were pretreated with serially diluted selinexor for 1 hour followed by a 4 hour stimulation with TNFα and then evaluated for the ability of the NF-κB p65 subunit to bind to a consensus binding sequence in a NF-κB activity assay. In this assay, selinexor was determined to have a NF-κB IC50 of 0.78 µM in NCI-H1299 cells and a somewhat higher IC50 of 1.19 µM in the less sensitive A549 cells (Figure 6A). In a subsequent western blot analysis, treatment with 0, 30, 100, 300, 1000 or 3000 nM selinexor for 24 hours decreased the protein levels of NF-κB p65 and IκB in both A549 and NCI-H1299 cells in a dose-dependent manner (Figure 6B). Selinexor could therefore be inhibiting activation of the NF- κB pathway by retaining IκB in the nucleus where it can neutralize NF- κB transcriptional activity, thereby preventing downstream pro-survival events in NSCLC cells.
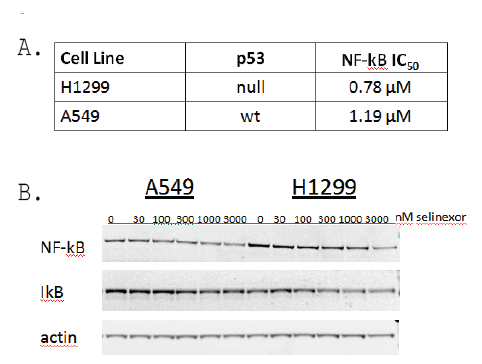
Figure 6: Selinexor modulated NF-ĸB activity and reduced protein expression of NF-ĸB and IĸB in A549 and NCI-H1299 cell lines regardless of p53 status. A) NF-ĸBIC50 determination from NCI-H1299 and A549 cells treated with 0.12, 0.37, 1.1, 3.3, 10, and 30 µM selinexor in serum-free media for 1 hour prior to the addition of 20 ng/ml TNFα for 4 hours. NF-ĸB IC50 values were calculated from total cell lysates evaluated for NF-ĸB activity in the NF-ĸB binding assay. B) Immunoblots of whole cell lysates from A549 and NCI-H1299 cells treated with 0, 30, 100, 300, 1000, or 3000 nM selinexor for 48 hours.
The effects of selinexor were next evaluated in vivo on the growth of the moderately sensitive NSCLC line A549 in a mouse xenograft model. Mice were subcutaneously inoculated with A549 cells and once the tumors reached a mean volume of ~100 mm3 , mice were treated as shown in Table 2. Selinexor showed dose-dependent inhibition of A549 xenograft growth, with maximal mean inhibition of 81% in the selinexor 20 mg/kg group compared to the vehicle group (Figure 7A) versus cisplatin with maximal inhibition of 13% compared to vehicle. Mean tumor volume changes were measured and reported in Figure 7B. This analysis indicated that there were statistically significant differences between the vehicle control group and the group treated with selinexor at 10 mg/kg (p=0.001) and 20 mg/ kg (p<0.0001).
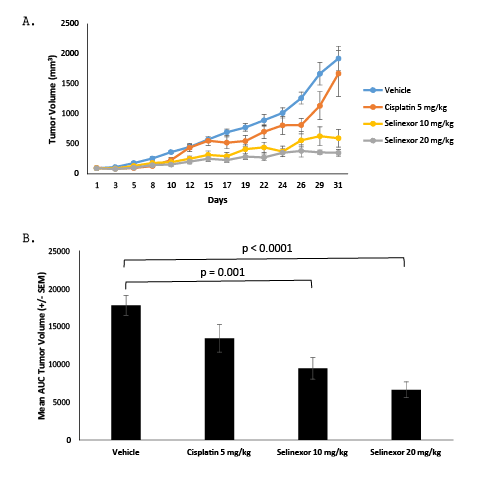
Figure 7: Treatment with selinexor reduced mean tumor volumes in an A549 xenograft model. A. Mean tumor volumes were calculated from the length and width measurements. Group means were calculated and are shown with error bars representing SEM for each group. B. The Area Under the Curve (AUC) was calculated using the trapezoidal rule transformation for the tumor volume measured from each animal in the study. Group means were calculated and are shown with error bars representing SEM for each group. Groups were compared using an ANOVA test, and statistically significant differences were seen between the vehicle control group and the groups treated with selinexor at 10 mg/kg (p=0.001) and 20 mg/kg (p<0.0001).
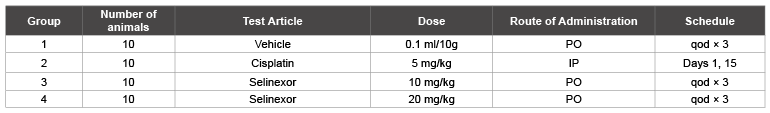
Table 2: A549 xenograft study groups
To better understand how selinexor inhibited tumor growth in the mouse xenograft model, tumors were harvested on day 31 and immunohistochemistry (IHC) performed. Representative images from each vehicle control and selinexor treated tumors are shown (Figure 8). Hematoxylin and eosin (H&E) staining showed the overall architecture of the tumors with and without selinexor (Figure 8A). Selinexor treated tumors showed more stroma (fibrotic) with fewer tumor cells than vehicle treated tumors. Treatment with selinexor reduced cell proliferation compared to the vehicle treated tumors as visualized by reduced Ki67 staining (Figure 8A). Although staining for p53 and p21 were not different in selinexor treated compared to vehicle treated tumors, selinexor-treated tumors showed increased nuclear accumulation of FOXO1 and survivin compared to vehicle treated tumors (Figure 8B). Since selinexor treatment induced nuclear localization of IκB, modulated NF-κB activity, and reduced NF-κB and IκB expression in NSCLC in vitro (Figures 4 and 6), the effect of selinexor treatment on this pathway was also evaluated in vivo. In tumors treated with selinexor, increased nuclear localization of both NF-κB and IκB was observed compared to vehicle treated tumors (Figure 8C). Nuclear co-localization of NF-κB and IκB is associated with inhibition of NF-κB activity [7,8], consistent with effects of selinexor on NF-κB in vitro (see above). Together these finding suggest that selinexor induces nuclear accumulation of proteins both in vitro and in vivo and modulates multiple pathways leading to NSCLC cell cycle arrest and death.
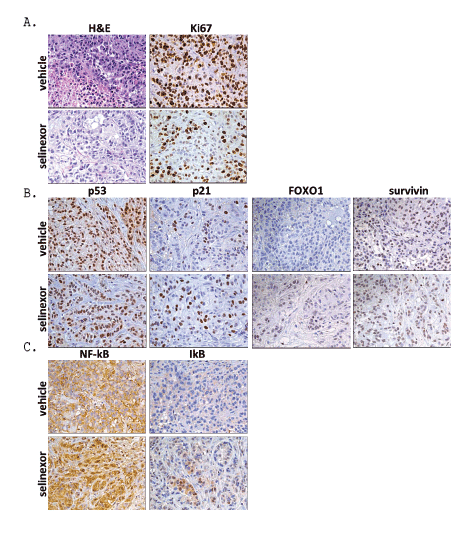
Figure 8: A549 xenograft histology. Selinexor inducedA) reduced proliferation (reduction in Ki67), B) nuclear accumulation of FOXO1 and survivin and
C) nuclear accumulation of NF-ĸBand IĸB (40x).
Supplementary Figure 1: NCI-H2030cells were resistant to selinexor at A) 10 µM but not at B) 60 µM. See figure 2 for a description of the cell cycle
profile analysis by flow cytometry. Resistant NCI-H2030 cells treated with the EC50 concentration of selinexor (10 µM) had decreased S phase with a
corresponding modest increase in G1 without significant cell death. By day 3, 60 µM selinexor caused death in all NCI-H2030 cells.
XPO1, a nuclear export protein and a member of the karyopherin-β family, has been shown to be overexpressed in a variety of different cancer types. Many studies have reported that inhibition of XPO1 nuclear export by SINE compounds results in nuclear accumulation of TSPs, inhibition of cell growth, and induction of apoptosis [10]. The clinical SINE compound, selinexor, is currently being evaluated in patients with advanced solid tumors, including lung cancer (NCT02351505), and thus far has demonstrated an acceptable safety profile and clinical benefit [24]. The preclinical data presented here support the anti-tumor activity and potential therapeutic effect of selinexor in NSCLC. In vitro, selinexor induced cell cycle arrest and apoptosis in a dose- and timedependent manner, induced cytoplasmic to nuclear localization of TSPs and cell cycle regulators, and modulated the NF-κB pathway regardless of histological sub-type and mutational profiles. in vivo, selinexor was a more effective treatment than cisplatin in a mouse xenograft model and showed histological profiles consistent with the in vitro data. These results demonstrate that selinexor is a potent and effective inhibitor of NSCLC.
This study directly interrogated whether in NSCLC the TSP p53 was required for the anti-cancer activity of selinexor. Consistent with observations reported with SINE compounds in other cancer types [22,30,32], selinexor exerted anti-cancer effects in NSCLC regardless of p53 status where similar results were seen in the p53 null NCI-H1299 and in the p53 wild type A549 cell lines. This suggests that although p53 has been shown to be important for cell cycle regulation and induction of apoptosis [33,34], it is not required for these events to be mediated by treatment with selinexor
Although the outcomes of treatment with selinexor on NCI-H1299 and A549 are similar, closer examination revealed distinct differences in their response to the compound over time, possibility reflected by their differing sensitivities to the drug. Comparison of the cell cycle profiles of each cell type when treated with selinexor revealed differences in accumulation of cells in the distinct phases of the cell cycle. The appearance of a larger sub-G1 fraction of apoptotic cells in NCI-H1299 compared to A549 cells was a reflection of the fact that NCI-H1299 cells are more sensitive to selinexor. The sub-G1 population was expected to increase in selinexor treated A549 cells over time. However, the increased G2 arrest in A549 cells that was not observed in NCI-H1299 cells indicated that each cell line responded differently to selinexor. Nonetheless, cell death was ultimately achieved in both cell types when treated with selinexor as evidenced by both the clonogenic assay as well as by western blot analysis for PARP cleavage and caspase activity.
Nuclear localization of most TSPs is required for their primary cell cycle checkpoint functions. Cancer cells overexpress XPO1 and other cofactors leading to the nuclear exclusion of key TSPs including p53, FOXO proteins, p27, and others that leads to functional inactivation of the TSPs, and contributes to resistance of neoplastic cells to apoptosis. Inhibition of XPO1 has previously been shown to lead to the accumulation of multiple diverse TSPs in the nucleus; export is prevented but their import from the cytoplasm is unaffected [12-15]. A hallmark feature of the effect of SINE compound treatment on cancer cells is the nuclear accumulation of many proteins involved in cancer progression [29,32,35-38]. Survivin is a member of the inhibitor of apoptosis family (IAP) and is highly expressed in a wide variety of cancers [39]. Survivin proliferative activity is exerted in the cytoplasm [40] and its forced nuclear retention may lead to its ubiquitination and proteasomal degradation [41]. Interestingly, patients with osteosarcoma, bladder, breast and lung cancers were reported to have favorable outcomes when survivin nuclear localization was observed [42,43]. The findings in this study demonstrated that selinexor-mediated nuclear entrapment of survivin in vitro and in vivo associates with cancer cell death.
In addition to survivin, selinexor treatment also induced the nuclear accumulation of the transcription factor E2F4. E2F4 is a cell cycle regulator that is exported to the cytoplasm by XPO1 to allow cells to enter into the cell cycle [44]. When confined to the nucleus, E2F4 binds to the three TSPs: p107, p130 and pRb and its binding is essential to activate their tumor suppression activities. Selinexor also induced pRB activity indicated by reduced nuclear detection of phospho-pRb (not shown) and it remains to be determined whether p107 and p130 are activated as well. Similar to survivin and E2F4, selinexor induced nuclear accumulation of p53 and its downstream target p21. p21 binds to and inhibits cyclinCDK2 and cyclin-CDK4 complexes and therefore arrests cells in the G1 phase of the cell cycle [45], as observed in the FACS analysis (Figure 2).
Although the mechanism is not fully understood, SINE compounds have been reported to increase nuclear localization of NF-κB and IκB as well as inhibit NF-κB activity in many different types of cancers [32,35,38]. The transcription factor NF-κB is normally bound to its inhibitor, IκB, in the cytoplasm [5]. Upon activation, NF-κB is released from IκB, which is targeted for degradation, and then NF-κB enters the nucleus to upregulate gene expression related to cell cycle progression as well as to inflammation [46]. In a negative feedback loop mechanism, IκB (itself an NF-κB regulated gene) enters the nucleus to prevent further NF-κBmediated gene activation [47]. Although NF-κB and/or IκB have yet to be definitively identified as XPO1 cargos, previous in vitro studies showed LMB treatment induced nuclear retention of NF-κB/IκB complexes and inhibited NF-κB activity which was attributed to the nuclear export signal identified in the N-terminus of IκBα [7,8]. In our study, selinexor inhibited the NF-κB pathway by inducing nuclear accumulation of IκB in vitro and in vivo, as well as decreased protein levels of NF-κB and IκB in both NSCLC cell lines, potentially resulting from a negative feedback loop on their own transcription. Inhibition of NF-κB activity occurred at lower concentrations of selinexor in the more sensitive NCI-H1299 compared to A549 cells, in agreement with increased baseline cytosolic expression/localization of IκB observed in A549 cells. The underlying mechanism of how selinexor abrogates the NF-κB pathway is currently under investigation.
Results from previous preclinical studies with SINE compounds in NSCLC cell lines were consistent with the findings in this study. Evaluation of the SINE compounds KPT-185 and KPT-276, early analogues of the clinical candidate selinexor, showed anti-cancer activity in NSCLC cell lines and mouse xenografts [22]. However, unlike the results reported here with selinexor, the previous study reported no effect on NF-κB or IκB expression. Although it resulted in reduced tumor growth, mice were treated with clinically irrelevant doses of the SINE compound KPT-276. Preclinical work with KPT-330 (selinexor) in NSCLC by Sun et al. [25] did not examine the NF-κB pathway nor investigate the effects of in vivo treatment of KPT-330 by histological analysis. In the present study, the cell cycle profile of the clinically-relevant XPO1 inhibitor selinexor was evaluated more extensively than the previous studies (3 days versus 24 hours), the NF-κB pathway was interrogated, clinically relevant doses of drug were tested in mouse models, and a thorough histological analysis of tumors treated with selinexor was performed. Regardless of the SINE compound tested or NSCLC cell lines evaluated, the data together consistently support SINE compounds having potent anti-cancer activity.
NSCLC, the predominant form of lung cancer, is characterized by poor prognosis and very limited treatment options. Survival rates are still low despite current targeted therapies for the treatment of NSCLC [4]. The effects of XPO1 inhibition by selinexor in vivo lead to the most exciting aspect of this study, which was the tremendous reduction in mean tumor volume in the A549 mouse xenograft model. The treatment was well-tolerated and showed significantly decreased mean tumor volume compared to the cisplatin positive control. Selinexor is currently being tested in Phase I and II/IIb clinical trials as both a single agent as well as in combination with other therapies to treat a variety of solid and hematological malignancies (Clinicaltrials.gov NCT01607892 and NCT01607905). The results from the Phase I clinical study as well as the in vivo data presented here suggest that the XPO1 inhibitor selinexor is a promising novel drug for NSCLC treatment.
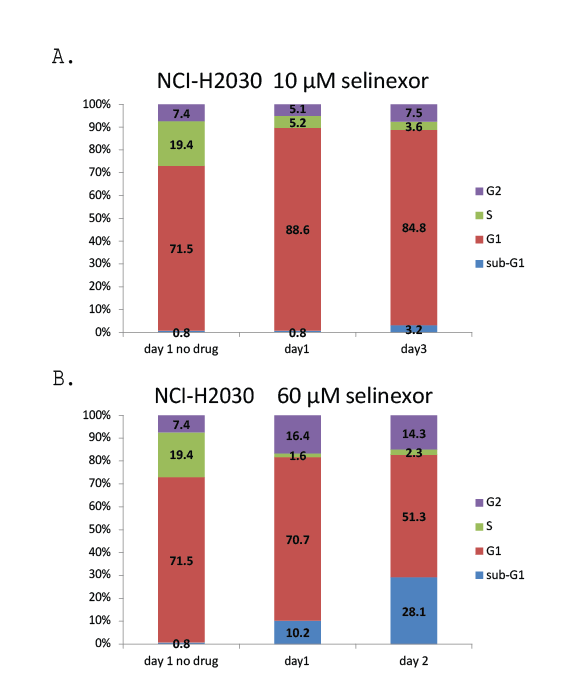
Supplementary Figure 1: NCI-H2030 cells were resistant to selinexor at A) 10 µM but not at B) 60 µM. See Figure 2 for a description of the cell cycle profile analysis by flow cytometry. Resistant NCI-H2030 cells treated with the EC50 concentration of selinexor (10 µM) had decreased S phase with a corresponding modest increase in G1 without significant cell death. By day 3, 60 µM selinexor caused death in all NCI-H2030 cells.
Download Provisional pdf here
All Sci Forschen Journals are Open Access