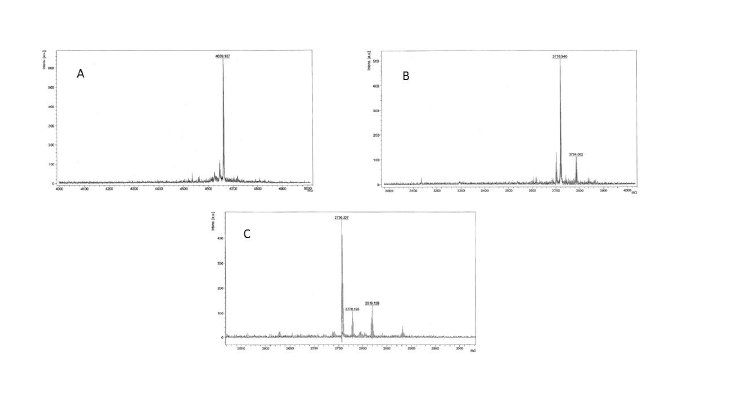
Figure 1: MALDI-ToF mass spectra of A) FSH33-53-Phor18; B) FSH81-95 -Phor18; C) FSH90-95-Phor18
Sita Aggarwal1* Ted Gauthier2 Hector Alila3 Carola Leuschner3 Namrata Karki1 Rajasree Solipuram1 Qingxia Wang1 William Hansel1
1Pennington Biomedical Research Center, Baton Rouge, LA, USA*Corresponding author: Sita Aggarwal, Pennington Biomedical Research Center, Baton Rouge, LA 70808, USA, Tel: 2257632931; E-mail: sita.aggarwal@pbrc.edu
Aritcle Type: Research Article
Citation: Aggarwal S, Gauthier T, Alila H, Leuschner C, Karki N, et al. (2015) Anti-tumor Effects of Targeted Follicle-stimulating Hormone-lytic Peptide Conjugates in Prostate Cancer (PC-3) Xenograft Mouse Model. Int J Cancer Res Mol Mech 1(4): doi http://dx.doi.org/10.16966/2381-3318.115
Copyright: © 2015 Aggarwal S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Conjugates of membrane disrupting lytic peptides with a 15-amino acid segment of the β chain of chorionic gonadotropin (CG) or luteinizing hormone releasing hormone (LHRH) target and destroy cancer cell xenografts in nude mouse model. Follicle stimulating hormone receptors (FSHR) have been detected in tumor neo-vascular endothelial cells of a variety of cancers and tumor tissue in prostate and ovarian cancers. In the present study, we have conjugated a lytic peptide (Phor18) to each of three segments of the β chain of FSH that bind to FSHR, and tested these conjugates (FSH90-95-Phor18, FSH81-95-Phor18 and FSH33-53-Phor18) for their ability to target and inhibit growth of prostate cancer cells in vivo. We found that intravenous administration of FSH90-95-Phor18, FSH81-95-Phor18 and FSH33-53-Phor18 significantly (p<0.05) inhibited the growth of prostate cancer (PC-3) xenografts in nude mice. The minimal effective dose of FSH81-95-Phor18 was 0.1 mg/kg and 1 mg/kg for FSH90- 95-Phor18 and FSH33-53-Phor18. Immunohistochemical analyses of tumor sections at necropsy showed dense expression of FSHR on vascular endothelial cells in control mice, reduced expression in tumors of mice treated with FSH90-95-Phor18, FSH81-95-Phor18 or FSH33-53-Phor18. FSH81- 95-Phor18 had the most potent anti-angiogenic activity of the three conjugates tested. These data show that a lytic peptide conjugated to FSH β chain segments bind to FSHR and are capable of inhibiting prostate cell tumor growth by targeting and destroying endothelial cells that express FSH receptors.
Prostate cancer; Follicle-stimulating hormone (FSH); Follicle-stimulating hormone receptor (FSHR); Lytic peptide; Vascular endothelial cells; Tumor growth inhibition; Anti-angiogenesis
Aside from non-melanoma skin cancer, prostate cancer is the most common cancer among men of all races. In the USA, prostate cancer is the second leading cause of cancer death in men and is associated with high morbidity and mortality [1]. The Cancer Statistics estimated new prostate cancer cases of 220,800 and 27,540 men died from the disease in 2015 [2]. Though currently used chemotherapeutic drugs are effective in reducing the mortality and morbidity caused by prostate cancer; but disease progression is inevitable [3-6]. One of the major problems associated with cancer treatment is dose-limiting toxicity that limits the doses of the chemotherapeutics that are systemically administered and do not specifically target the cancer cells. These facts led to attempts to develop more effective drugs that are able to specifically target cancer cells. It is widely accepted that luteinizing hormone/human chorionic gonadotropin (LH/hCG) and LHRH receptors are highly expressed in various tumor cells. Several lytic peptide drug conjugates that target LH/hCG and LHRH receptors expressed by prostate, breast, ovarian, pancreatic and testicular cancer cells have been tested in our laboratory in the past few years [7,8].
Among these drugs is a class of membrane disrupting peptides [9,10] that are abundantly present in nature. Tumor cells are 50 times more sensitive to these membrane-disrupting peptides than normal cells [11], suggesting their usefulness in destroying various cancers. In previous reports, we showed that conjugates of membrane disrupting lytic peptides with either luteinizing hormone releasing hormone (LHRH) or with a 15-amino acid segment of the β chain of hCG target and destroy human prostate cancer cells, breast cancer cells, ovarian cancer cells and pancreatic cancer cells in tumor bearing nude mice [12,13]. A LHRH receptor targeting membrane disrupting peptide conjugate, EP-100, has been developed by Esperance Pharmaceuticals and completed Phase 2 clinical studies [14].
Like LH, follicle-stimulating hormone (FSH) is a key gonadotropin hormone in mammalian reproduction. FSH is produced mainly in the anterior pituitary gland, and the target organs are the ovary and testis. FSH is a heterodimer consisting of a common α-subunit non-covalently associated with β-subunit. All mammalian β subunits of FSH have 111 amino acids. FSH receptor is a glycosylated transmembrane protein that binds FSH and belongs to the family G-protein-coupled receptors. In adult humans and animals, the FSH receptor is expressed only in the testicular Sertoli cells and the ovarian granulosa cells [15,16] and in low levels in the endothelial cells of the ovary [17] and testis [18]. Recently, it has been reported that the FSH receptor is expressed in vascular endothelial cells of at the periphery in a wide range of human tumors, including prostate cancer [19]. Two previous studies confirmed the presence of FSH receptors in prostate adenocarcinoma by immunohistochemical detection [20,21]. The role of FSH as a marker of extra prostatic extension in human prostate cancer is significant. Therefore, these results suggest that FSH receptor and its ligand FSH may play an important role in prostate carcinogenesis [22].
Given these recent reports and earlier findings from our laboratory showing effectiveness of lytic peptide conjugates in inhibiting tumor progression, we conceptualized that development of conjugates of lytic peptides with FSH could be an effective approach to target and destroy human prostate cancer cells. To date, a number of lytic peptide conjugates have been synthesized and their activities have been tested [8,13]. One such membrane-disrupting lytic peptide is Phor18, an 18-amino acid peptide that was developed at Esperance Pharmaceuticals for targeting LHRH receptors in cancers by conjugating Phor18 to LHRH (EP-100). EP-100 has been successfully administered intravenously to 76 patients, and demonstrated activity and safety [23]. In the present study, we synthesized a bio conjugate of Phor18 to each of three segments of the β chain of FSH that are known to bind to the FSH receptor. The three segments of the β chain of FSH used were 6 amino acids (90-95), 15 amino acids (81-95) and 21 amino acids (33-53) resulting in three conjugates FSH90-95-Phor18, FSH81-95-Phor18 and FSH33-53-Phor18. Subsequently, we assessed the ability of all three conjugates to target and lyse prostate cancer xenografts and assessed their effects on tumor growth and neovasculature.
A comprehensive understanding of the molecule’s characteristics, including its destruction of prostate metastases in animal models is important for further development of FSH-Phor18 as a novel therapeutic option.
Binding segments of β-FSH conjugated to a lytic peptide (phor18) were; amino acids, 90-95 (Asp-Ser- Thr-Asp-Cys-Thr); amino acids, 81-95 (Gln-Cys-His-Cys-Gly-Lys-Cys-Asp-Ser-Asp-Ser-Thr-Asp-CysThr); amino acids, 33-53 (Tyr-Thr-Arg-Asp-Leu-Val-Tyr-Lys-Asp-ProAla-Arg-Pro-Lys-Ile-Gln-Lys-Thr-Cys-Thr-Phe). All peptides were synthesized using standard fluorenylmethylcarbonyl (Fmoc) solid phase chemistry on a Protein Technologies Tribute automated synthesizer. In short, each conjugate was synthesized on pre-loaded Wang resin using 4.85 equivalents of 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3- tetramethylaminium hexafluorophosphate (HCTU), 10 equivalents of 4-methylmorpholine (NMM) and 5 equivalents of Fmoc amino acid. Couplings were allowed to proceed for 20 minutes followed by 1-methyl- 2-pyrrolidinone (NMP) washes (5x, 30 sec). Fmoc removal was achieved with a solution of 20% piperidine in NMP (2.5min) followed by NMP washes (5x, 30 sec). The peptide was simultaneously cleaved from the resin and side-chain deprotected using 95% trifluoroacetic acid, 2.5% water and 2.5% triisopropylsilane. In the cases where thiol containing amino acids were present in the peptide, the cleavage cocktail was modified to include 94% trifluoroacetic acid, 2.5% water, 2.5% ethanedithiol, and 1% triisopropylsilane. Peptides were purified by preparatory scale HPLC with a Waters prep LC controller and 2489 UV/Vis detector. The separation was performed on an AAPPTEC Spirit Peptide 120 C18 column (5 µm, 21.2 × 250 mm) with a Spirit Peptide guard column (21.2 × 10 mm). Solvents A (0.1% TFA in water) and B (0.1% TFA in acetonitrile) were mixed in a gradient starting at 5% B to 55% B in 50 minutes. The flow rate was 20 mL/ min and the detected wavelength was 215 nm. Purity of each of the FSHlytic peptides was checked by single peaks obtained on a Waters analytical HPLC equipped with an AAPPTEC Spirit Peptide 120 C18 column (5 µm, 4.6 × 250 mm) and a Spirit Peptide guard column (3.2 × 10 mm). The solvents and gradient were identical to those used in the prep HPLC. The MALDI-Tof mass spectrum for each peak in the analytical HPLC verified the identity of each peptide. The purity was >95% for all three conjugates.
Human prostate cancer cells (PC-3) were obtained from American Type Culture Collection (ATCC) VA, USA and cultured according to the ATCC protocol. PC-3 cells were cultured in Dulbecco Modified Eagle Medium (DMEM) supplementedwith 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 µg/ml streptomycin (Invitrogen, CA, USA).
FSH receptor (323) monoclonal antibody was kindly supplied by Dr. Ghinea, INSERM, France.
Ethics: All animals used in these investigations were handled in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and the Guidelines of the US Department of Agriculture (Animal Welfare Act; Public Law 99-198). The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee at the Pennington Biomedical Research Center. Male nude mice (athymic Balb/c, nu/nu, 5 weeks old) obtained from Charles River Laboratories, Wilmington, MA, were housed in standard mouse Plexiglas cages in a room maintained at constant temperature and humidity under a 12 h light and darkness cycle. Animals were fed irradiated pelleted chow (PicoLab Rodent Diet 20, Ren’s Feed and Suppliers Ltd., Oakville, ON) with water ad libitum.
Subcutaneous implantation of PC-3 cells: PC-3 cells were harvested from subconfluent cultures after a brief exposure to 0.25% trypsin and 0.2% EDTA. Trypsinization was stopped by adding medium containing 10 % FBS. The cells were washed once in serum-free medium and resuspended in PBS. Only suspensions consisting ofsingle cells with >90% viability were used for the injections. PC-3 cells(1 × 106 ) in 100 µL PBS suspended in Matrigel (0.1 ml) (Collaborative Biomedical Products Becton Dickinson Labware, Bedford, MA) were injected subcutaneously into the interscapular area using a 27-gauge needle. On day 21, after tumor cell implantation, mice bearing tumors between 100-200 mm3 , were randomized into the following treatment groups (n=8); Vehicle controls; FSH90-95 peptide (1 mg/kg), FSH81-95 peptide (1 mg/kg), FSH33-53 peptide (1 mg/kg), FSH90-95-Phor18 (0.1 mg/kg, and 1 mg/kg), FSH81-95- Phor18 (0.1 mg/kg, and 1 mg/kg), and FSH33-53-Phor18 (0.1 mg/kg, and 1 mg/kg) treatments. Peptides and peptide conjugates were dissolved in USP saline and further diluted with saline to achieve a dose concentration of 1 mg/kg (0.2 mg/ml) dose and 0.1 mg/kg (0.02 mg/ml) dose. Volumes of 100 µL were given intravenously by tail vein injections twice weekly and given five injections in total. The vehicle treated group received an equivalent amount of USP saline. Tumor volumes were measured with calipers twice a week and body weights of mice were measured once a week. Tumor volumes were calculated by the formula: V (mm3 )=(length × width2 )/2, in which width was the shortest measurement in millimeters. Mice were necropsied one week after the last treatment on day 36th. At necropsy, tumors were dissected, weighed, photographed and portions were fixed by immersion in 5% neutral buffered formalin. The histology tissue samples were processed into paraffin blocks, and 5 μm sections were cut for further studies.
Immunoperoxidase staining was performed for FSH receptors on sections from the tumor tissue samples obtained from nude mouse xenografts. Sections were deparaffinized with xylene (3 times for 10 min. each) and rehydrated through ascending ethanol concentrations (30%, 50%, 70%, 95% and 99.7–100%) followed by an antigen retrieval step. In this procedure, slides were placed in boiling 0.01 M sodium citrate and left standing at room temperature for a further 35 minutes to unmask epitopes. The slides were then rinsed in phosphate buffer saline (PBS) twice for 5 min each. Endogenous peroxidase activity was inactivated by immersing slides in 1% (v/v) hydrogen peroxide in methanol for 20 min. The slides were washed twice for 5 min each in PBS. Subsequently, sections were blocked for 60 min in a humidified chamber using a blocking solution, comprised of 1% normal horse serum (Vectastain Universal Elite ABC kit, Vector Laboratories, CA, USA), diluted in PBS and 20% (v/v) avidin solution (Avidin/Biotin blocking kit; Vector Laboratories, CA, USA). After washing in PBS (3 times for 10 min each), the slides were incubated overnight at 4℃ in a humidified chamber with the FSH receptor (323) monoclonal antibody at dilution of 1:50. The primary antibodies were diluted in PBS to which 20% (v/v) biotin solution (Avidin/Biotin blocking kit; Vector Laboratories, CA, USA) was added. The negative control sections were treated in the same manner with PBS and biotin mixture in an absence of primary antibodies. After, the slides were washed thoroughly with PBS (3 times for 10 min.) and incubated for 30 min. with the secondary antibody (Biotinylated anti-mouse IgG Vector Laboratories, CA, USA). After another rinse for 10 min., the avidin–biotin complex solution (Vectastain Universal Elite ABC kit, Vector Laboratories, CA, USA) was applied for 30 min. After incubation, slides were washed twice for 5 min each in PBS. Tissue sections were then incubated with NovaRED peroxidase substrate (NovaRED; Vector Laboratories, CA, USA). Slides were then counterstained with Hematoxylin (DAKO, USA) for 30 sec., and washed with distilled water and with descending ethanol concentrations (100%, 95%, 70%, 50%, and 30%) and soaked in xylene for 5 min. Slides were then mounted with a drop of clear mount (Electron Microscopy Sciences, USA). FSH receptors were stained as a red-brown color; negative controls showed no staining.
Double immunoperoxidase staining was performed for FSH receptors and anti-von Willebrand factor, a specific marker of vascular endothelial cells, in sections from the tumor tissue samples obtained from nude mouse xenografts. The preparation of samples was conducted as described above including antigen retrieval and blocking. The slides were incubated overnight at 4℃ in a humidified chamber with the FSH receptor (323) monoclonal antibody at a dilution of 1:50 and the rabbit polyclonal antivon Willebrand factor (AbCam, USA), a specific marker of vascular endothelial cells at a dilution of 1:50. The negative control sections were treated in the same manner with PBS and biotin mixture in the absence of primary antibodies. The slides were then washed thoroughly with PBS (3 times for 10 min.) and incubated for 1 hour with a mixture of goat antirabbit Alexa 594 (Molecular Probes, USA) and goat anti-mouse Alexa 488 (Molecular Probes, USA) secondary antibodies. In the same experiment, nuclei were detected by incubating slides for 15 min. with Hoechst stain (Sigma, USA). The slides were mounted with a drop of clear mount (Electron Microscopy Sciences, USA). Pictures were taken with Leica Zeiss Confocal Laser Scanning Microscope at 63X with water emersion.
All results were expressed as mean ± S.E. unless otherwise noted. Statistical significance was assessed by Student’s t-test or one-way analysis of variance was applied to compare the means of two or more groups simultaneously, p<0.05 was considered to be statistically significant.
Mass spectra are shown in Figure 1(A-C). Purity of each of the FSHPhor18 conjugate was obtained on mass spectra. The purity was >95% for all three conjugates.
Results of the in vivo experiment, in which doses of 0.1 mg/kg, and 1 mg /kg of each of the three FSH-Phor18 conjugates were tested for their abilities to inhibit growth of PC-3 xenografts in nude mice are shown in Figures 2(A-C) and 3(A-C). The data illustrate that administration of FSH90-95-Phor18 and FSH81-95-Phor18 significantly (P<0.05) inhibited prostate cancer cell growth in vivo, as measured by tumor weights Figure 2(A-C) and tumor volumes Figure 3 (A-C) at necropsy when compared with vehicle group. The effective dose for FSH90-95-Phor18 was 1 mg/kg body weight, while the effective dose for FSH81-95-Phor18 was 0.1 mg/kg. The FSH35-53-Phor18 conjugate caused a significant reduction of tumor volume at necropsy at a dose of 1 mg/kg, but the reduction in tumor weight was not statistically significant when compared with vehicle group. None of the segments of the β chain of FSH had any effect on tumor weight or volume when administered alone and compared with vehicle group.
To further investigate the effects of the lytic peptide conjugates on prostate tumor development, we measured tumor volume twice every week. As depicted in Figure 4 (A-C), vehicle treated mice bearing prostate tumor xenografts had a time-related increase in tumor volume. FSH90-95- Phor18, FSH81-95-Phor18 and FSH33-53-Phor18 caused significant reduction in tumor growth at the dose of 1 mg/kg from day 25 when compared to vehicle treated group and each of the FSH-peptide fragment treated groups. No side effects in FSH-Phor18 treated groups were found as no changes were found in liver and kidney function test in treated groups (data not shown). There was no significant difference in the mice weight at necropsy between the vehicle and treated groups (Figure 4(E-F)).
We next examined whether FSH-Phor18 conjugates reduce FSH receptors on the neovasculature of the tumors. To examine this, immunohistochemical analysis was performed on paraffin-embedded tissue sections by the use of the anti-FSH-receptor monoclonal antibody (323), followed by a secondary peroxidase-coupled antibody visualized with the use of the red-brown peroxidase-reaction product. Sections were stained with hemotoxylin. When compared to tumors of vehicle treated (Figure 5A), fewer FSH-receptor-positive vascular endothelial cells were present in tumors of nude mice treated with FSH90-95-Phor18 (Figure 5B) FSH81-95-Phor18 (Figure 5C), and FSH33-53-Phor18 (Figure 5D).
Stained vessels are indicated by solid arrows; unstained vessels by open arrows. No staining was observed when the immunohistochemistry was performed on tissue sections without primary antibody (Figure A. Supplementary data).
In order to confirm the presence of FSH receptors on the neovasculature of the tumors, we stained paraffin embedded sections of tumor xenografts using double immunofluorescence staining. We stained the sections using antibodies against FSH receptor (green) and von Willebrand factor (vascular endothelial cell marker; red). FSH receptors were co-localized with von Willebrand factor, as indicated by yellow fluorescence in the merged images (Figure 6; panel D). Hoechst stained nuclei are shown in Figure 6; Panel A. These results clearly show the presence of FSH receptors in vascular endothelial cells of the prostate tumor xenografts in nude mice. Negative controls (Figure B. Supplementary data).
The goal of the current study was to synthesize conjugates of the membrane disrupting lytic peptide (Phor18) and one of the three segments of the beta chain of FSH (6 to 21 amino acids) and to test each of these conjugates for their potency in reducing tumor volume and weight in a FSH receptor positive prostate cancer xenograft model and it’s effect on the neovasculature of treated tumors. The salient findings of the study were: 1) Administration of FSH90-95-Phor18, FSH81-95-Phor18, and FSH33- 53-Phor18 significantly inhibited prostate cancer cell (PC-3) growth and volume; 2) FSH81-95-Phor18 was found to be effective in reducing primary tumor weight and volume in nude mice bearing human tumor xenografts at lower dose levels than FSH90-95-Phor18, FSH33-53-Phor18; 3) this present study provides further evidence of the presence of FSH receptors in vascular endothelial cells and tumor cells of the prostate tumor xenografts in nude mice; and 4) Fewer FSH-receptor-positive vascular endothelial cells were present in tumors of nude mice treated with FSH81-95-Phor18, FSH90-95-Phor18 and FSH33-53-Phor18 than in tumors of vehicle treated mice. This study is the first to demonstrate the development of these conjugates of a lytic peptide and FSH and testing their effectiveness in destroying prostate tumor cells in vivo. These findings clearly show the ability of conjugates consisting of the membrane disrupting lytic peptide, Phor18 and selected FSH segments to inhibit the prostate tumor growth in nude mice bearing human prostate tumor xenografts. FSH81-95-Phor18 was the most potent conjugate of the three FSH-Phor18 conjugates tested.
Figure 1: MALDI-ToF mass spectra of A) FSH33-53-Phor18; B) FSH81-95 -Phor18; C) FSH90-95-Phor18
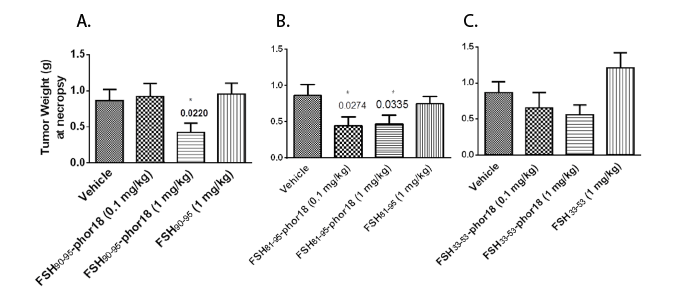
Figure 2(A-C): Tumor weights at necropsy-The data represent mean ± s.e. (n=8). Tumor weights at necropsy were significantly reduced in FSH90-95-Phor18 and FSH81-95-Phor18 treated mice compared to vehicle treated groups. * p <0.05.
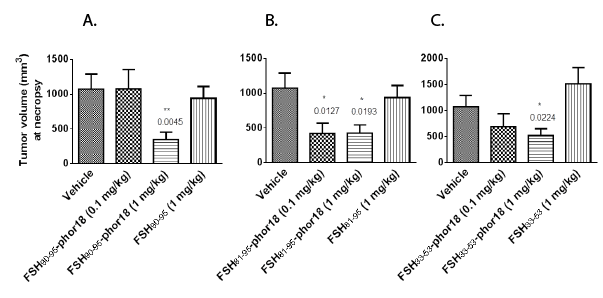
Figure 3(A-C): Tumor volumes at necropsy-The data represent mean ± s.e. (n=8). Tumor volume at necropsy was significantly reduced in FSH90-
95-Phor18 and FSH81-95Phor18 and FSH33-53-Phor18 treated mice compared to vehicle treated groups.
*
p<0.05.
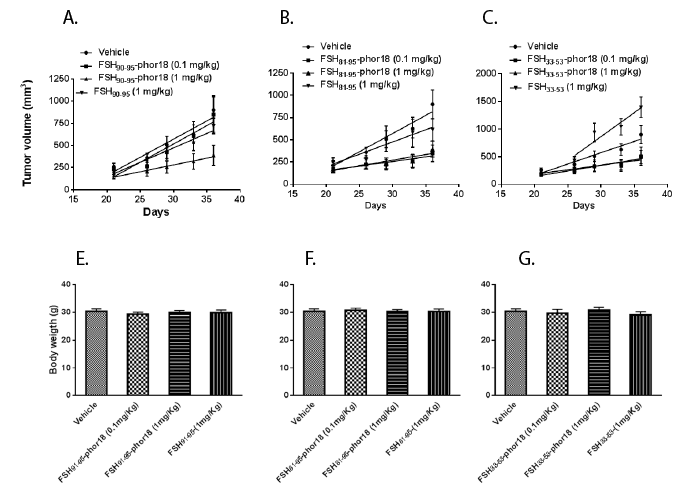
Figure 4(A-C): FSH-Phor18 conjugates inhibit the growth of human prostate cancer cells in vivo -Changes in tumor volumes in nude mice
treated with normal saline (vehicle controls), FSH 90-95 (1 mg/kg), FSH90-95-Phor18 (0.1 mg/kg, 1 mg/kg ), FSH81-95 (1 mg/kg), FSH81-95-Phor18 (0.1 mg/
kg, 1mg/kg ), and FSH33-53 (1 mg/kg), FSH33-53-Phor18 (0.1 mg/kg, 1 mg/kg ) treatments. Data are presented as the means ± s.e. of tumor volume (n=8).
4(E-F): Body weights of animals treated with FSH-Phor18 conjugates-There were no significant differences between body weights of vehicletreated
and FSH-Phor18 conjugates treated animals.
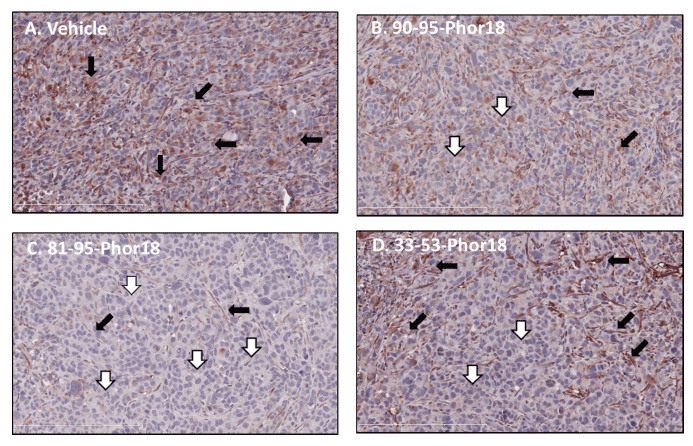
Figure 5: Expression of follicle stimulating hormone (FSH) receptors by blood vessels in human prostate cancer cell xenografts after treatment with FSH-Phor18-Immunohistochemical analysis was performed on paraffin-embedded sections of human prostate cancer cells grown in nude mice by the use of the anti-FSH-receptor monoclonal antibody (323), followed by a secondary peroxidase-coupled antibody visualized with the use of the red-brown peroxidase-reaction product. Sections were stained with hemotoxylin. Fewer FSH-receptor-positive vessels were present in tumors of nude mice treated with FSH81-95-Phor18, FSH90-95-Phor18 and FSH33-53 -Phor18 than in tumors of vehicle treated mice (compare Panel A to Panels B, C and D). FSH81-95-Phor18 appears to be the most potent of the three FSH-Phor18 conjugates tested. Stained vessels are indicated by solid arrows; unstained vessels by open arrows.
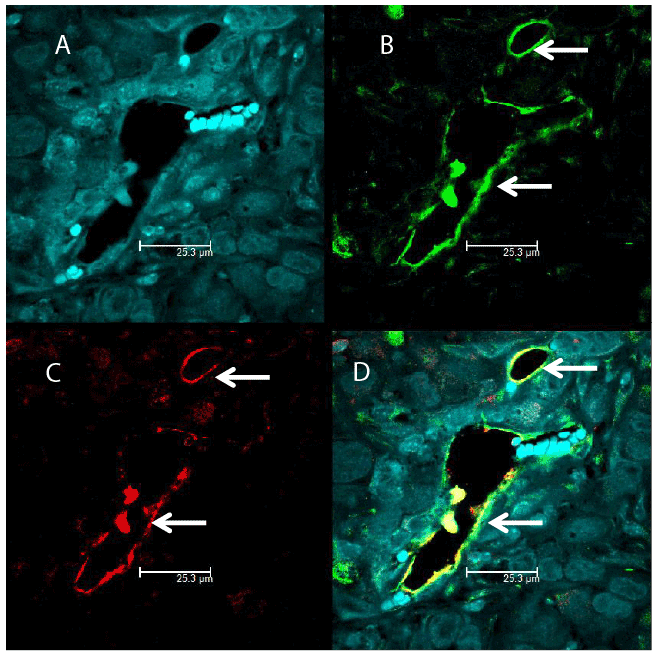
Figure 6: Identification of endothelial cells in prostate tumor cells expressing FSH receptors by double immunofluorescence-An antibody against the vascular endothelial cell marker von Wille brand factor, followed by a red-labeled secondary antibody (Panel C) overlapped with the signal from the anti-FSH-receptor antibody determined by a secondary green labeled antibody (Panel B). Merging of the two antibody signals is shown in Panel D (yellow color). Hoechst stained nuclei are shown in Panel A. Picture were taken at 63X with 1.2 Aperture.
Additionally, our results demonstrate that the anti-tumor effects of these conjugates are mediated by specific binding to FSH receptors expressed on the blood vessels supplying the prostate tumors and on FSH receptors on tumor cells. Our results are in agreement with previous studies that have demonstrated that FSH receptors selectively expressed on the surface of the blood vessels of a wide range of human tumors including prostate cancer [19-21].
Non tumoral tissues and their vasculature of pleura, lung, liver, bone, and lymph nodes do not express FSHR [19]. Several markers have been described that are preferentially expressed on blood vessels in tumors (e.g., prostate specific membrane antigen, αv β3 -integrin, vascular endothelial growth factor and its receptors) and in the extracellular matrix surrounding newly formed blood vessels (matrix metalloproteinases, Robo-4) [24]. However, as suggested previously, targeting FSHR of the tumor neovasculature and tumor cells could enhance the potency of the FSH-Phor18 in killing tumors and can be more effective than targeting other known markers because many of these markers (e.g. integrins) do not allow highly specific targeting of tumors [25]. It has recently been reported that FSHR levels in primary renal cell carcinoma tumors correlate strongly with the response of metastatic tumors in patients treated with sunitinib, an antiangiogenic receptor tyrosine kinase inhibitor suggesting a direct relation between FSHR levels and extent of metastasis [26,27]. Our current results are significant from a clinical perspective because FSHR are absent in most healthy tissues; hence, targeting FSHR could minimize the damage that anti-cancer drugs do to surrounding tissues or organs. The lytic peptide conjugates have the added advantage that they are not antigenic because they are small and are metabolized with minimal side effects such as, probable inhibition of spermatogenesis in males and ovulation in females [28]. A limitation of our study is increased risk for animal’s fertility. However, it was previously shown that the function of the pituitary gonadotrophs was completely recovered within 2 weeks after treatment with a targeted cytotoxic analog of LHRH [29]. Furthermore, the recombinant FSH treatment restored serum inhibin-B levels and partially restored spermatogenesis in gonadotropin-deficient rats [30].
The present study demonstrates the effectiveness of the FSHPhor18 conjugates against primary prostate tumors through targeting neovasculature and tumors. Because the lytic peptides used in the present study are specifically bound by the targeting segment (βFSH), we suggest that our FSH-Phor18 conjugates would not only destroy primary tumors but also metastatic tumors. Reinforcing this concept, previous reports from our laboratory have shown that lytic peptides bound with LH or hCG prevent metastasis. Nonetheless, it would be interesting to investigate whether these conjugates are equally effective in targeting metastatic tumors, and these conjugates would overall increase survival rate of the treated mice, could be a focus for future studies. Taken together, these results suggest that FSH-Phor18 conjugates could be novel therapeutic options for the treatment of prostate cancers in humans.
We thank, Dr. David Burk for assistance with fluorescence microscopy. The Bioimaging Core at Pennington Biomedical Research Center is supported, in part by COBRE (NIH P20-RR021945) and CNRU (NIH IP30-DK072476) and the Pennington Biomedical Research Foundation. We thank Dr.Ghinea, INSERM, France for providing FSH receptor (323) monoclonal antibody.
This work was supported by Esperance Pharmaceuticals, Inc., Baton Rouge, LA; and by the Hansel/Downey Research Fund, the Pennington Biomedical Research Foundation and by Louisiana State University, LA.
Download Provisional pdf here
All Sci Forschen Journals are Open Access