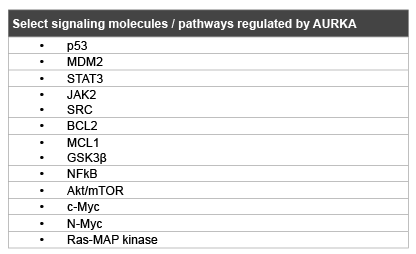
Table 1: Summary of key molecules interacting with AURKA
Pokuri VK Opyrchal M Boland PM*
Department of Medicine, Roswell Park Cancer Institute, Elm & Carlton Streets, Buffalo, NY 14263, USA*Corresponding author: Boland PM, Assistant Professor, Department of Medicine, Roswell Park Cancer Institute, Elm & Carlton Streets, Buffalo, NY 14263, USA, E-mail: Patrick.Boland@ RoswellPark.org
Aritcle Type: Review Article
Citation: Pokuri VK, Opyrchal M, Boland PM (2015) Aurora Kinase A and Gastrointestinal Malignancies. Int J Cancer Res Mol Mech 1(3): doi http://dx.doi. org/10.16966/2381-3318.114
Copyright: © 2015 Pokuri VK, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Aurora kinase A (AURKA) has emerged as a potential therapeutic target in gastrointestinal malignancies. It has been shown to not only regulate the process of mitosis, but also to have critical non-mitotic function. AURKA is involved in epithelial to mesenchymal transition and upregulation of stem cell-like cells. The AURKA gene has been localized to chromosome 20q13. Changes in expression levels of AURKA are frequently detected in human neoplasms including various GI malignancies. Aberrations of aurora kinase signaling can lead to dysregulation of cell growth, altering the activity of a growing number of partner proteins and pathways. In pre-clinical work, AURKA has been shown to play a role in carcinogenesis, propagation of tumors and resistance to chemotherapy in GI malignancies. Despite pre-clinical promise, initial clinical studies have yielded only limited successes. Novel agents and combinations, based upon an increasing understanding of AURKA’s role, offer potential for new therapeutic anti-cancer strategies. This article represents a brief review of AURKA and its role in neoplasia, as well as a review of the clinical activity of various compounds in patients with GI malignancies.
Aurora kinase; Gastrointestinal cancer; Targeted therapy
Aurora kinases (AURK/Ipl1p family) are a family of highly conserved serine/threonine protein kinases (A, B and C) that play a vital role in mitosis and/or meiosis [1]. The Aurora A and B (AURKA and B) were originally described as regulators of mitosis in cell cycle studies of model organisms like Xenopuslaevis (African clawed frog) [2-4], Drosophila melanogaster (fruit fly) [5,6] and Caenorhabditis elegans (nematode) [7,8]. Subsequent studies in mammals identified the third Aurora kinase C (AURKC) apart from the other two [9]. These kinases have different subcellular localizations and functions. AURKA (STK15/BTAK) is localized to centrosomes and mitotic spindle poles/microtubules and thus regulates mitotic entry, centrosome maturation and bipolar spindle assembly (Figure 1) [10,11]. AURKB is localized to the kinetochores at the centromeres during early phases of mitosis and acts as a chromosomal passenger protein, serving in chromosome condensation, orientation and cytokinesis [11,12]. AURKC is similar to AURKB in localization and function; however, it is mainly expressed in gonads, regulating spermatogenesis and oocyte development (meiosis) [11,13,14]. Due to the importance of these molecules in the regulation of mitosis and chromosomal trafficking, aberrant expression of members of the aurora kinase family leads to genomic instability.
AURKA is the most extensively studied of the Aurora kinase family members. It is coded by a gene, Aurora A, located on chromosome 20q13, initially called Breast Tumor Activated Kinase (BTAK) as it was found to be overexpressed in transformed human breast cancer cell lines [15]. AURKA was found to be upregulated in colorectal malignancies as well as many other malignancies, including ovarian, prostate, neuroblastoma and cervical cancer cell lines [1,16]. It has been found that TPX2 and IQGAP1 proteins result in increased AURKA stability and their overexpression has been shown to promote oncogenesis [17-19]. Conversely, decreasing expression of CHFR, which results in degradation of AURKA, has also been implicated in oncogenesis [20]. Overexpression of AURKA causes deregulation of centrosome duplication, distribution, chromosome segregation and cytokinesis resulting in aneuploidy [1,21]. Upregulated AURKA was also shown to phosphorylate p53 at serine315, causing its degradation via Mdm2 dependent ubiquitination [22]. Thus, there is a direct link between AURKA and dysregulation of p53. Collectively, AURKA induced alterations are capable of down-regulating cell cycle check-point pathways and promoting oncogenic transformation.
AURKA has also been proposed to interact with multiple other pathways of importance in GI malignancies. AURKA has been linked to control of STAT3 activity, with overexpression of AURKA enhancing STAT3 phosphorylation and nuclear translocation [23]. Conversely, inhibition of AURKA via siRNA as well as specific inhibitors results in decreased phosphorylation and transcription of STAT3, mediated through JAK2. Potentially as a consequence of STAT inhibition, the antiapoptotic targets, BCL-2 and MCL1, are also downregulated via AURKA inhibition in multiple tumor types, including gastrointestinal cancers [23,24]. Several other mechanisms through which increased expression of AURKA could promote cancer progression were described, increased NFκB transcription factor activity [25], increased activity of Akt pathway in a p53-dependent manner [26], increased oncogenic Ras signaling [27], increased expression of c-Myc [28] and N-Myc [29] (Table 1).
Table 1: Summary of key molecules interacting with AURKA
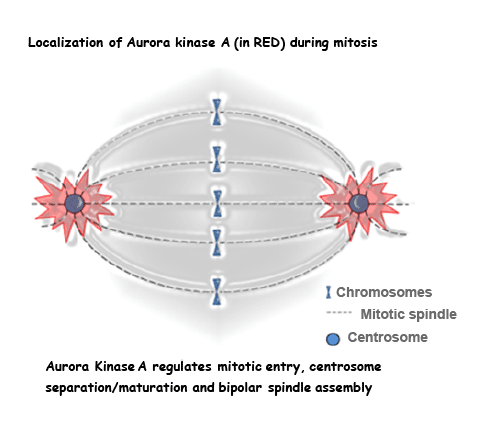
Figure 1: AurkA and the mitotic spindle
AURKA has been implicated in inducing resistance of cancer cells to standard treatments [26,30,31]. In various models, Aurora kinase inhibitors have been shown to have increased activity in combination with chemotherapy [32-34], underscoring the potential of aurora kinase inhibition to overcome chemo-resistance in cancer cells. One of the proposed mechanisms has been that AURKA leads to cancer progression and metastasis through induction of epithelial to mesenchymal transition (EMT) and increased cancer cell population with stem celllike characteristics [35,36]. EMT and the acquisition of stem cell-like properties have been linked to resistance to standard treatment regimens [37]. In a colorectal cancer model, knockdown of AURKA resulted in growth inhibition of colorectal cancer stem cells, with decreased tumorigenic potential as demonstrated by decreased ability to engraft and form viable tumors in immunocompromised mice. AURKA knockdown has also resulted in decreased metastatic potential of the cancer cells [38]. These studies highlight the oncogenic role of AURKA in malignant transformation, tumor progression and resistance to current therapies.
Prognostic significance of AURKA expression has been studied in gastrointestinal cancers. In colorectal carcinoma (CRC), studies evaluating the prognostic significance of AURKA expression have shown conflicting results, though most demonstrate that increased expression of AURKA correlates with worse clinical outcomes. Several studies showed a strong association between accumulation of gains in 20q13, the site of AURKA among other putative oncogenes, and adenoma-to-carcinoma progression in colorectal cancer [39-42]. These data highlight the potential mechanistic role for AURKA as one of the driving forces of carcinogenesis. Gains of 20q13.2 (by fluorescence in situ hybridization), where in one series were identified in 53% of sporadic CRC patients, correlate with faster tumor progression and worse patient survival, independent of tumor size and lymph node involvement [43]. In this data set, 20q13 gain was inversely correlated with tumor grade and associated with left-sided colon cancers.
Multiple retrospective studies have looked at protein expression of AURKA or gene copy number. In one study, including 200 patients with CRC of various stages, AURKA protein expression by immunohistochemistry (IHC) was noted in 48.5% of samples, more frequently in those with well or moderately differentiated colorectal carcinomas, as well as left sided tumors [44]. There was a trend toward worse outcomes with AURKA expression, not reaching statistical significance. A second group examined 517 CRC cases, stages I-IV, and identified AURKA overexpression by IHC in 19% of patients. A significant association between AURKA overexpression and chromosomal instability was observed, which was not related to site of origin. However, in this analysis, AURKA overexpression was not significantly associated with survival [45]. On the other hand, in an analysis of 386 patients with stages II-III colon cancer, a slightly more uniform population, higher AURKA expression by IHC was associated with higher risk of disease recurrence [46]. In the metastatic setting, 61 patients were examined for AURKA gene copy number, with 68% having increased gene copy number by real time PCR [47]. Contrary to the prior data sets, the increase in AURKA was associated with improved overall and progression free survival in this population. Only a limited sub-set of patients were able to undergo IHC based analysis. Finally, in yet another analysis of metastatic patients, 343 colorectal liver metastasis resection specimens were analyzed for AURKA expression by IHC [48]. Expression within the hepatic lesions correlated with that seen in the primary tumors. Importantly, when comparing the 30% of patients with strong expression to those with lesser expression of AURKA, significantly worse survival was observed. This difference was maintained upon multivariate Cox regression analysis, which included established clinic opathologic predictors of outcome. In summary, while there have been differences in methodology, most studies correlate AURKA expression to worse prognosis, particularly when looking at larger data sets and those with a more homogenous patient population.
In non-colorectal gastrointestinal cancers, a number of studies have highlighted the poor prognostic impact of AURKA protein overexpression or gene amplification. Greater than 50% of upper GI cancers demonstrate overexpression of AURKA by IHC [49]. Elevated AURKA expression in primary duodenal adenocarcinoma is predictive of poorer overall survival in multivariate regression analysis [50]. In gastric cancers, AURKA expression by IHC appears to correlate with nodal involvement, lymphovascular invasion, and advanced stage [51]. In multivariate analysis, AURKA overexpression remains an unfavorable predictor of survival. Another study demonstrated upregulation of Aurora kinase A mRNA by RT-PCR in 30% and protein expression in >50% of esophageal squamous cell carcinomas [52]. Upregulation by either methodology correlated with the presence of distant lymph node metastasis and poorer prognosis. Again, on multivariate analysis protein expression remained an independent negative prognostic variable for survival. In a study of resectable gastroesophageal cancer patients who underwent neoadjuvant chemo-radiation and surgical resection, analysis of single nucleotide polymorphisms (SNPs) in the coding region of AURKA (STK15) was performed [53]. The Phe31/IIe variant of T91A was associated with worsened odds of recurrence, as well as inferior survival. An additional SNP was evaluated, with the presence of at least one variant allele at each locus markedly increasing risk of recurrence (adjusted OR=6.21). Limited further validation of these data sets has been published. In completely resected primary gastrointestinal stromal tumors (GISTs) and metastatic GISTs, AURKA overexpression was identified as an unfavorable prognostic marker with worse recurrence-free survival and overall survival [54,55]. In summary, AURKA expression, by various methods, appears to be adversely linked to patient outcomes in GI cancers.
The aurora kinase inhibiting compounds can be divided into AURKA selective or pan-Aurora kinase inhibitors. Inhibition of aurora kinases has shown promising pre-clinical activity in gastrointestinal tumors of various origins. R1498, a multi-kinase inhibitor whose target includes Aurora kinases, showed significant antitumor activity in xenograft models of gastric and hepatocellular carcinomas [56]. Another group demonstrated the antitumor activity of the pan-aurora kinase inhibitor Danusertib (PHA-739358) in an orthotopic xenograft model of metastatic gastroenteropancreatic neuroendocrine tumors [57].
In vitro and in vivo studies utilizing the selective oral first generation AURKA inhibitor, MLN8054, resulted in proliferation arrest in multiple human tumor cell lines including human colorectal cancer xenografts; mechanistically this appeared to occur via mitotic accumulation and apoptosis, resulting in phenotypes consistent with Aurora A inhibition [58]. In combination with docetaxel, Alisertib (MLN8237) resulted in significant antitumor activity in upper gastrointestinal adenocarcinoma cell lines and xenograft models. Anti-tumor activity was observed irrespective of p53 status, inducing aberrant mitosis, polyploidy and apoptosis [59]. Alisertib has also exhibited promising antitumor activity in combination with cisplatin in esophageal adenocarcinoma cell lines, as well as docetaxel in gastric and esophageal models [59,60]. Finally, a novel aurora-A inhibitor, BPR1K0609S1, was shown to sensitize human colon carcinoma cell lines and xenograft models to 5-fluorouracil [61].
The exact mechanism for chemosensitization is unclear. AURKA’s role as a mitotic checkpoint represents one possible explanation. An alternative hypothesis focuses on the non-mitotic function of AURKA and its ability to induce EMT and increase the percentage of cells with stem cell-like characteristics; both of which have been associated with resistance to chemotherapy agents in GI malignacies. Studies have demonstrated inhibition and reversal of EMT with AURKA signaling blockade. In both pancreatic and gastric cancer cell lines, compounds which inhibit AURKA induce cell cycle arrest and inhibit and reverse the process of EMT [62,63]. Through a series of experiments which examined populations of CD133+ colorectal cancer stem-cell like cells, AURKA inhibited Twist and Snail expression (two genes closely linked to EMT), markedly diminishing the migratory capacity of cancer cells. Inhibition of AURKA further increased the chemosensitivity of this relatively resistant cell population to a multitude of agents, including5-FU and oxaliplatin [38].
Combined targeting of AURKA and the EGFR-RAS-MAPK pathway has seen some preclinical benefit. A synthetic lethality screen identified AURKA as a target which might synergize with anti-EGFR inhibition. Initial experiments supported this avenue of investigation [64]. In melanoma cell lines, AURKA inhibitors demonstrated marked activity when combined with MEK and/or BRAF inhibitors [65]. More recently, in colorectal cell lines and xenografts, the combined inhibition of MEK with TAK-733 and AURKA with alisertib was studied; double KRAS/PIK3CA mutant cells lines demonstrated synergistic inhibition of proliferation with the combination treatment as compared to either treatment alone [66]. Beyond the EGFR-MAPK pathway, alisertib has been combined with inhibitors of KIT and ABL (e.g. imatinib) where synergy was exhibited in GIST cell lines [55].
Multiple aurora kinase inhibitors have been developed (Table 2), with many undergoing the initial process of clinical development: single agent testing in phase I studies. MLN8054 was studied in several phase I trials, producing initial signals of durable antitumor activity in multiple tumor types. In a phase I study of MLN8054 in 61 patients, 22 of whom had refractory colorectal cancer, 9 patients (15%) achieved stable disease for at least 4 cycles of treatment [67]. No complete or partial responses were seen. One colorectal cancer patient in this study received the minimum dose of 5 mg orally daily for 7 days ( and off for 14 days, constituting a 21 day cycle) and experienced stable disease lasting for 8 cycles of treatment. The dose limiting toxicity (DLT) was somnolence, predicted based upon structural similarity to benzodiazepines. Unfortunately, despite alternate dosing schedules or use of psychostimulants (e.g. methylphenidate or modafinil) this toxicity proved insurmountable [67]. An additional study produced similar results: disease stabilization in a minority of patients, with the DLT of transaminitis [68]. Very few patients with other gastrointestinal cancers were included, precluding any assessment of clinical benefit. Pharmacokinetic/pharmacodynamic studies of MLN8054 revealed that at the maximum tolerated dose, Aurora A inhibition was incomplete. Given marked toxicity, which precluded further escalation and adequate target inhibition, further development was halted.
At present, Alisertib is the AURKA inhibitor which is furthest in clinical development, correspondingly with the greatest amount of clinical data. Two phase I studies of Alisertib in advanced solid cancers showed similar responses of stable disease over 3 to 6 months in 10-23% patients [69,70]. Among the gastrointestinal cancers in these studies, most were colorectal cancers (42 patients) with others being pancreatic (5), anal (5), gastroesophageal (3) and gall bladder (2). In these initial phase I studies, 1 patient with metastatic colorectal cancer experienced stable disease for greater than 12 months. The most common dose limiting toxicities of Alisertib were neutropenia and stomatitis, while fatigue, nausea and neutropenia were the most common adverse events in these studies [69,70]. The recommended phase 2 dose of Alisertib was 50 mg orally twice daily for 7 days in a 21 day cycle [70,71]. This dose is currently being studied in several phase 2 clinical trials.
Recently, the results of a multi-armphase II study of single agent Alisertib in advanced, heavily pretreated solid tumors were reported. Patients were selected for tumors which have been shown to overexpress AURKA and included breast cancer, small cell lung cancer, nonsmall cell lung cancer, head and neck squamous cell carcinoma and gastroesophageal adenocarcinoma [72]. The toxicity profile was similar to that seen in the phase 1 trials; neutropenia was the most common grade 3-4 adverse event [72]. Alisertib demonstrated differential activity in this trial, with a response rate (RR) of 18% and 21% in breast and small cell lung cancers, respectively, while only 9% (4/47 patients) of patients with gastroesophageal adenocarcinomas experienced responses. Due to the small fraction of patients experiencing benefit, the median progression free survival (PFS) was uninspiring in gastroesophageal adenocarcinoma (1.5 months).
Danusertib is a pan-aurora kinase inhibitor which was studied in a phase 1 dose escalation study of patients advanced refractory solid tumors. The established the maximum tolerated dose was 500 mg/m2 , given as 24 hour intravenous infusion every 2 weeks [73]. Nineteen colorectal cancer patients were involved in this study and prolonged disease stabilization (23.9-52.3 weeks) was observed only in two of them (11%). An additional phase II study utilizing this dosing schema was conducted in heavily treated patients with advanced solid tumors [74]. The safety profile of Danusertib was similar to prior studies and neutropenia was the most frequent adverse event. This study showed only marginal activity in GI cancers with a progression free survival of 4 months in10% (3/30 patients) in pancreatic cancers and 0% (0/28 patients) in colorectal cancers.
AT9283 is an AURKA and B inhibitor which has been investigated in the phase I setting. DLT was found to be neutropenia. At MTD there was evidence of target inhibition and modest clinical activity was observed. Though there were no RECIST responses, best clinical response observed was stable disease seen in 1/6 (17%) esophageal cancer patients and in 1/12 (8%) colorectal cancer patients.
MSC199237A is a pan-aurora kinase inhibitor, which has seen investigation as a single agent, as well as in combination with gemcitabine. In the monotherapy phase I study, cytopenia and fatigue represented the most common grade 3/4 toxicites [75]. Again, no partial or complete responses were achieved. Stable disease was achieved in a substantial proportion of evaluable patients (41%). This included one patient with a neuroendocrine tumor who experienced minor response (17% reduction in tumor burden) of 18 months duration. An additional study of MSC199237A in combination with gemcitabine was conducted [76]. This drug proved tolerable with standard doses of gemcitabine (1000 mg/m2 ), with expected neutropenia as DLT. There were only 2 patients with confirmed partial responses (3%). However, several patients with GI cancers saw some clinical benefit. One out of 6 patients with hepatocellular carcinoma (HCC) experienced a partial response, remaining on study for 11 cycles. One patient of two (50%) with esophagogastric cancer attained stable disease lasting over 9 months, and one additional patient of 8 (13%) with pancreatic cancer also achieved disease stability lasting greater than 7 months.
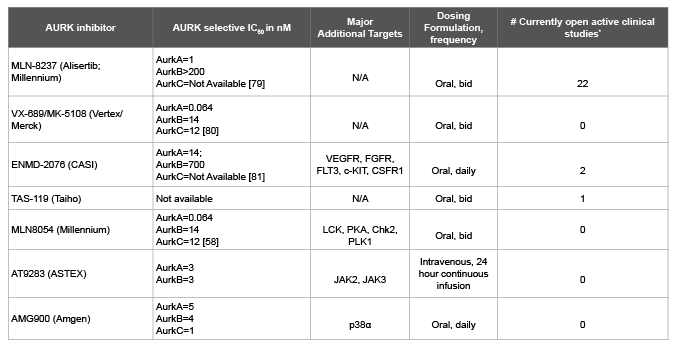
Table 2: Profiles of Select Aurora Kinase Inhibitors
There are multiple ongoing, completed and planned studies of aurora kinase inhibitors in combination with chemotherapy. The AURKA inhibitor, Alisertib, has seen the greatest use in this setting. Tolerability has been demonstrated with weekly paclitaxel as well as docetaxel with some signs of clinical benefit [77]. Recently, data was presented on alisertib combined with gemcitabine utilizing an alternate schedule: days 1-3, 8-10, and 15-17 of a 28 day cycle. In a study of multiple tumor types (most commonly NSCLC), there were 18 evaluable patients, with 1 (6%) patient experiencing a PR and 10 (56%) experiencing stable disease as best response. Though there was a significant incidence of grade III/IV hematologic toxicity, the vast majority of patients received full doses of both drugs [78]. An expansion cohort of patients with pancreatic cancer is ongoing and will be of great interest. Multiple combinatorial investigations are ongoing across tumor types with studies of particular interest targeting GI tumors highlighted in Table 3.
Aurora kinase inhibitors, with alisertib being most advanced, are currently being evaluated in multiple phase I and II trials, alone and in various combinations. As single agents, their efficacy appears to be limited in gastrointestinal tumors with best responses predominantly being stable disease. Given that alisertib demonstrated promise in preclinical investigations of upper GI malignancies when added to chemotherapeutics and demonstrates evidence of single agent clinical efficacy, further investigation is merited. Alternate schedules might be worthy of investigation, due to class-related toxicity of myelosuppression. With gemcitabine, which has the potential to induce significant myelotoxicity, a slight adjustment in dosing strategy allowed for the addition of alisertib with reasonable tolerability at full doses and some signs of benefit.
Though in the small phase II study of alisertib in gastroesophageal cancers the median PFS was limited, robust responses were observed, suggesting that there may be clinical benefit for select patients. Notably, preclinical activity has been demonstrated when AURKA inhibition is combined with fluoropyrimidines, platinum compounds and taxanes, across multiple tumor types, particularly in gastric and esophageal cancer cell lines. These three classes of agents are instrumental in the treatment of advanced gastroesophageal cancers. In the future, such combinations may be tested. There is intensive interest in combining AURKA inhibitors with hypothesis driven targeted agents with non-overlapping toxicities. JAK2-STAT3 pathway activation by AURKA was recently found to be an important driver of carcinogenesis in upper GI malignancies; more extensive work is needed to determine if these combinations have future in clinical development. Pre-clinical data suggest potential in exploring combination therapies with Aurora inhibition together with inhibitors of EGFR-RAS-MAPK pathway. Additional work is needed to identify patient population which may benefit from this approach.
In order to fully establish AURKA inhibition as viable treatment option for patients, development of reliable clinical predictors of efficacy is needed. Ongoing and future studies will hopefully identify prospective biomarkers of clinical benefit or lack of thereof, which will have to be tested in a prospective manner. Additional pre-clinical work fully delineating non-mitotic function of AURKA will help to develop innovative correlative studies.
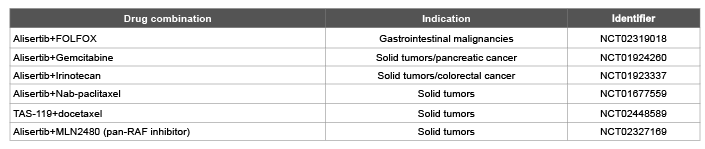
Table 3: Ongoing Clinical Studies of Interest Utilizing Aurora Kinase Inhibitors
Although the preclinical data of AURKA inhibition in gastrointestinal cancers is promising, the initial clinical activity of this class of compounds in patients with GI malignancies has been modest at best. Pharmacokinetic and pharmacodynamic testing has suggested that AURKA inhibition in tumor tissue is attainable at MTD. More work is needed to identify the optimal partners for combination therapies and biomarkers for predicting therapeutic benefit. We are eagerly awaiting the results of the ongoing clinical trials to determine if inhibitors of AURKA will have a role in treatment of patients with GI malignancies.
Download Provisional pdf here
All Sci Forschen Journals are Open Access