Introduction
Cisplatin, cis-diamminedichloroplatinum (II), is a well-known
chemotherapeutic drug and is one of the most widely used drugs for the
treatment of various cancers [1,2]. Cisplatin is clinically proven to combat
different types of cancers including sarcomas, cancers of soft tissue, bones,
muscles, and blood vessels. Recent developments in the chemotherapy
of such cancers have yielded better prognosis and therefore have led to
these diseases becoming less life threatening [3]. From the molecular
perspective of cisplatin, it represents a perfect example of how a small
alteration in chemical structure can significantly affect biological activity
in target cells. Nine platinum analogs are currently in clinical trials around
the world [4,5].
Cisplatin is known to bind cellular components such as DNA and
proteins, and to form DNA and protein adducts as a result of cross links
with DNA and protein molecules that hamper transcription and translation
mechanisms [6,7]. Those DNA adducts impact cell cycle progression check
points and determine whether the cell has to die or survive with the help
of repair mechanisms [8]. In addition to the formation of DNA adducts,
cisplatin induces oxidative stress, and modulates calcium signaling, and
cell apoptosis [9,10]. It also regulates the expression of many proteins that
play a major role in cell cycle regulation and signal transduction [8,11,12].
Cisplatin also mediates the mitogen activated protein kinases (MAPK)
and jun amino-terminal kinase (JNK) pathways that coordinate various
extra cellular signals to regulate cell growth, survival, and apoptosis [13,14].
Additionally, cisplatin induced oxidative stress may trigger cell death
besides DNA damage. Oxidative stress is one of the important processes
that induce toxicity by targeting the mitochondrion membrane potential,
and eventually leads to the inhibition of calcium uptake due to loss of
mitochondrial protein sulfhydryl group. The degree of oxidative stress
induction is dependent on the exposure time and drug dose [15]. The
thiol group (– SH) containing molecules play a major role by maintaining
the intracellular redox homeostasis. Reactive oxygen species are generated
when thiol radicals interact with molecular oxygen under induced
conditions [3]. If the cells cannot control the generation of reactive
oxygen species, they may eventually damage cellular membranes and
trigger intrinsic and/or extrinsic pathways of apoptosis [16]. However,
in contrary to entering the apoptotic pathway in some - patients, cells
develop resistance to cisplatin due to a decrease in drug intake or a
passively diffusion of drug [17,18].
Although some of the biochemical effects of cisplatin are well studied,
the detailed mode of its potential therapeutic action at low doses has not
yet been elucidated. The goal of present research was to assess the low
dose effects and elucidate the cytotoxic mechanisms of cisplatin on HL-60
cells. Our results provide a scientific basis to identify the lowest dose of
cisplatin that has a maximum impact in reducing cancer cell proliferation,
thereby minimizing its side effects on normal/non-cancer cells. Hence,
in this study, we have investigated the cytotoxicity, oxidative stress, lipid
peroxidation and genotoxicity potentials of cisplatin at low levels of
exposure to HL-60 cells.
Methods and Materials
Cell culture, chemicals and reagents
Cisplatin was obtained from University of Mississippi Medical Center
(Jackson, MS). Iscove’s Modified Dulbecco’s Medium (IMDM) and
HL-60 cells were purchased from American Type Culture Collection
(ATCC) (Manassas, VA, USA). Fetal bovine serum (FBS), and penicillin
– streptomycin were purchased from Sigma (St. Louis, MO, USA). IMDM
contains 4 mM L-glutamine, 4500 mg/L glucose, and 1500 mg/L sodium
bicarbonate, and is supplemented with 10% (v/v) FBS, and 1% (W/V)
antibiotics. The live cells were incubated at 37°
C in a 5% CO2 incubator
(Thermoscientific, Waltham, MA, USA).
Cell treatment
The experiments were conducted with three replicates for each
control or treatment group. Uniform cell density of 1 × 106
cells/mL was
maintained for all the treatments and controls. Cells were treated with 1,
2, or 3 µM of cisplatin or left untreated, and incubated for various time
periods (24, 48, 72, or 96 h) at 37°C in a humidified 5% CO2
incubator
(Thermoscientific, Waltham, MA, USA).
Cell proliferation assay
Cell viability was assessed as previously described [19] with CellTiter
96®
AQueous One Solution Cell Proliferation Assay kit from Promega
(Madison, WI, USA) with few modifications. Briefly, 100 µL aliquots of the
treated or untreated cell suspension were seeded into 96-well polystyrene
tissue culture plates and 20 µL of assay reagent was added to each well.
After 60 min of incubation at 37°C, the absorbance was read at 490 nm
with a 96 well plate reader from BMG LABTECH GmbH (Ortenberg,
Germany).
Lipid peroxidation assay
Malondialdehyde (MDA) concentrations were quantified as previously
described [20] using lipid peroxidation assay kit (Abcam, Cambridge, MA,
USA) with few modifications. After the each treatment, cell suspension
was collected, centrifuged at 1,230 rpm for 5 min to get the cell pellet and
the cell pellet was suspended in cell lysis buffer. After centrifugation at
13,000g for 10 min, a 200 μl aliquot was assayed for MDA levels according
to the lipid peroxidation assay kit protocol. The absorbance of the sample
was read at 532 nm with a 96-well plate reader from BMG Labtech GmbH
(Ortenberg, Germany). The concentrations of MDA were determined
from the standard curve.
Superoxide dismutase (sod) and catalase assays
SOD and Catalase assays were carried out as previously described
[21,22] with few modifications, using commercially available SOD assay
kit and Catalase assay kit (Abcam, Cambridge, MA, USA), respectively.
After each treatment, both the control and treated HL-60 cells were
collected. Cell lysates were quantified for SOD and catalase activities
according to the manufacturer’s protocol. The final absorbance for SOD
was measured at 450 nm and catalase was measured at 570 nm using 96-
well plate reader from BMG Labtech GmbH (Ortenberg, Germany).
Single cell gel electrophoresis
Cisplatin genotoxicity in treated and untreated HL-60 cells was
analyzed by alkaline single cell gel electrophoresis (Comet) assay as
described earlier [23] with few modifications using Comet assay kit for
single cell gel electrophoresis from Trevigen (Gaithersburg, MD, USA).
All the precautions were taken to avoid the UV light effect on DNA. Low
melting agarose was melted in boiling water and cooled down to 37°C.
For each treatment, 75 μL of the agarose and cells mixtucre (ratio of 1:10)
was placed on comet slides and solidified at 4°C. Cells were lysed with
lysis solution for 30 min at 4°C, followed by denaturing the DNA with
alkaline solution for 40 min at 37°C. The prepared slides were subjected to
electrophoresis (1 volt/cm) for 10 min in TBE (Tris borate EDTA) buffer.
After the electrophoresis, cells were fixed with 70% ethanol followed by
staining with SYBR green. Epifluorescent microscope (Olympus BX51
TRF, USA) was used to observe the comet slides. The data was evaluated
using the DNA damage analysis software (Loats Associates Inc., USA).
Statistical analysis
Experiments were carried out in triplicates, and the data were presented
as means ± SDs. To test for differences among and between experimental
groups, one–way analysis of variance (ANOVA) and Student’s t-test were
performed respectively, using SAS software available in the Biostatistics
Core Laboratory available at the RCMI Center for Environmental Health
at Jackson State University for testing differences. Data were considered
statistically significant for p-values less than 0.05.
Results
Cisplatin inhibits HL60 cell proliferation
Cell survival was measured in HL60 cells treated with 1, 2, or 3 µM
cisplatin for 24, 48, 72, and 96 h incubation periods compared to the
respective controls (Figure 1). The results indicate that the cell viability
was decreased with increased cisplatin dose and incubation period. For
1 µM cisplatin, the percentages of cell viability were 92.0 ± 1.7%, 81.0 ±
3.8%, 59.0 ± 2.2% and 38.0 ± 1.9% for 24, 48, 72, and 96 h treatment,
respectively. The recorded data for 2 µM cisplatin were 89.0 ± 2.2%, 74.0
± 4.0%, 52.0 ± 3.9% and 41.0 ± 2.8% for 24, 48, 72, and 96 h treatment,
respectively. The cell viability percentages for 3 µM cisplatin were 84.0 ±
3.1%, 66.0 ± 1.8%, 48.0 ± 3.3%, and 34.0 ± 3.6% for 24, 48, 72, and 96h
treatment, respectively. Compared to the 1 µM treatment; cell survival
rates decreased by about 8%, 15%, 11%, and 4% in 3 µM cisplatin, for
24, 48, 72, and 96h treatment, respectively. Overall, significant dose- and
time-dependent decreases (p<0.05) were observed in cisplatin-treat cells
compared to control cells.
Cisplatin elevates SOD and catalase activities in HL60 cells
SOD and catalase activities were estimated in untreated or treated
HL60 cells with 1, 2, or 3 µM cisplatin for 24, 48, 72, and 96 h time
periods. The results were presented in Figures 2 and 3 for SOD and
catalase, respectively. SOD and catalase activities were increased as the
cisplatin dose or time period increases. At the lowest concentration, 1 µM
cisplatin, SOD activity was increased by 23%, 30%, 62%, and 82%, while
catalase activity was increased by 29%, 33%, 41%, and 58% compared to
their respective controls at 24, 48, 72, and 96 h of exposure, respectively.
In 2 µM cisplatin-treated cells, SOD activity was increased by 41%, 47%,
67%, and 86% while catalase activity was increased by 37%, 39%, 40%,
and 57% compared to their respective controls, at 24, 48, 72, and 96 h of
exposure, respectively. At the highest concentration, 3 µM cisplatin, SOD
activity was increased by 61%, 68%, 73%, and 110% while catalase activity
was increased by 41%, 45%, 49%, and 75% compared to their respective
controls at 24, 48, 72, and 96h of exposure, respectively. The increases in
SOD and catalase activities in cisplatin-treated cells were significantly
different from the respective controls (p<0.05).
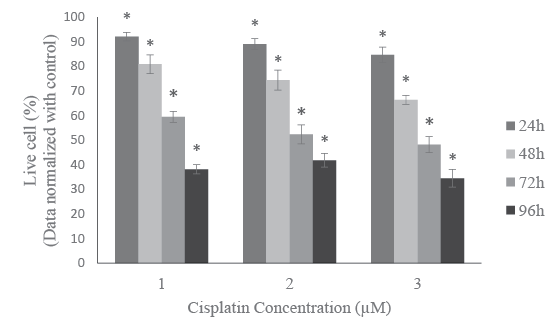
Figure 1: Cytotoxic effect of cisplatin on HL-60 cells. Cells were treated
with 1, 2 and 3 µM of cisplatin for 24h, 48h, 72h and 96h. Cell viability
was tested by cell proliferation assay using a spectrophotometer at 490
nm. Data wasnormalized with control to one. Results were expressed
as means of three independent experiments ± standard deviations.
p-values less than 0.05 were considered statistically significant.
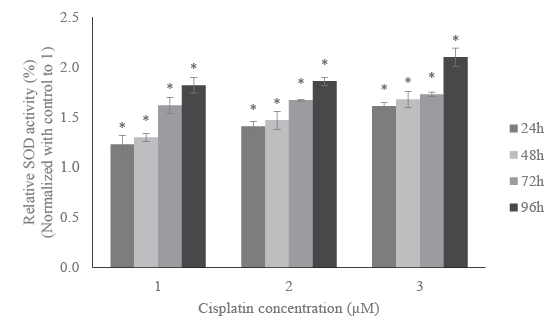
Figure 2: Effect of cisplatin on SOD activity in HL-60 cells. SOD activity
was measured for the HL-60 cells challenged with 1, 2 and 3 µM of
cisplatin for 24h, 48h, 72h and 96h. Post treatment, cells were lysed
and SOD activity was quantified. The absorbance of each sample was
measured at 450 nm and results were presented in the graph. Data
was normalized with control to 1 and expressed as means of three
independent experiments ± standard deviations. p-values less than
0.05 were considered statistically significant.
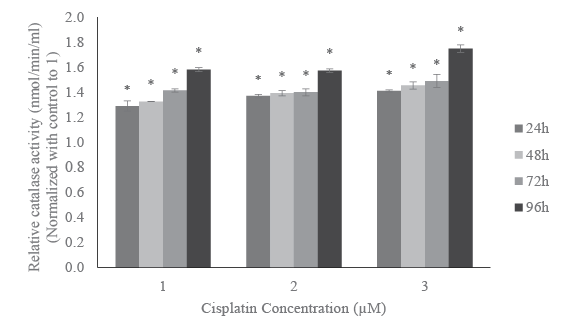
Figure 3: Effect of cisplatin on catalase activity in HL-60 cells. HL-60
cells were incubated with 1, 2, and 3 µM of cisplatin for 24h, 48h, 72h,
and 96h. After each treatment, catalase activity was estimated using
spectrophotometer at 570 nm. Results were expressed in nmol/min/ml.
Data was normalized with control to 1. Data were expressed as mean
of three independent experiments ± standard deviations. p-values less
than 0.05 were considered statistically significant.
Cisplatin induces lipid peroxidation in HL60 cells
One of the best methods for assessing oxidative stress is the estimation
of MDA, a byproduct of lipid peroxidation. The MDA concentrations were
estimated in HL-60 cells treated with cisplatin. The data are presented in
Figure 4. As shown on this figure, the estimated MDA levels gradually
increased in a dose- and time- response manner. After 96 h of exposure
HL-60 cells treated with 1, 2 and 3 µM cisplatin induced 39, 48, and 64%
more MDA compared to the respective controls. A similar trend was also
found for any other exposure time periods. Hence, the increased MDA
levels for all the tested concentrations and time periods were significantly
higher than the respective controls (p<0.05).
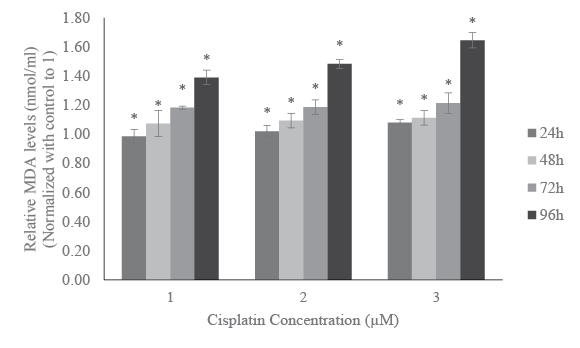
Figure 4: Effect of cisplatin on lipid peroxidation in HL-60 cells. HL-60
cells were treated with different doses of cisplatin or left untreated for
24h, 48h, 72h, and 96h. MDA levels were estimated in the treated and
untreated cells. The amount of MDA (nmol/ml) was measured at 532
nm. Data was normalized with control to 1. The results were expressed
as mean of three independent experiments ± standard deviations.
p-values less than 0.05 were considered statistically significant.
Cisplatin induces DNA damage in HL60 cells
Treatment of HL60 cells with cisplatin at doses 1, 2 or 3 µM for a period
of 24, 48, 72, and 96 hour shows dose- and time-response relationships
with regard to DNA damage. Figure 5 presents the representative Comet
assay images of HL-60 cells showing substantial increases in DNA damage
at higher levels of cisplatin exposure. These qualitative data were analyzed
quantitatively based on three replicates using the DNA damage analysis
software (Loats Associates Inc., USA), The quantified DNA damage data
presented in Figure6 indicated that HL-60 cells treated with 3 µM cisplatin
for 96 h induced the highest level of DNA damage, whereas those treated
with 1 µM cisplatin for 24 h induced lowest level of DNA damage. At 1
µM, the percentages of DNA damage were1.6 ± 0.12%, 2.8 ± 0.51%, 3.6 ±
0.47%, and 5.7 ± 0.23% for 24, 48, 72, and 96 h exposure, respectively. At 2
µM these percentages were 3.4 ± 0.19%, 3.60 ± 0.38%, 6.1 ± 0.46% and 7.1
± 0.28% for 24, 48, 72, and 96 h exposure, respectively. At the highest dose,
3 µM of cisplatin, the percentages of DNA damage were 5.9 ± 0.09%, 6.5
± 0.12%, 6.7 ± 0.39%, and 10.5 ± 0.16% for 24, 48, 72, and 96 h exposure,
respectively. The induced DNA damage is significantly higher for all the
tested concentrations compared to the respective controls (p<0.05).
Discussion
The present study was carried out to investigate the cytotoxicity,
oxidative stress, and genotoxicity of cisplatin in HL60 cells. The main
aim of this study was to research the anti-cancer potential of low doses of
cisplatin, in order to minimize effect on non-cancer cells. We evaluated
cell viability for cytotoxicity, anti-oxidants levels and lipid peroxidation
for oxidative stress, and comet assay for genotoxicity.
Cisplatin is a neutral inorganic chemical compound that induces
cytotoxicity by characteristic inhibition of cell cycle progression as a result
of DNA adducts formation and subsequent induction of apoptosis. To
exhibit toxicity, cisplatin has to hydrolyze with water molecules to become
an active molecule that interacts with various macro molecules in the cell
[17,24].The reactive cisplatin activity is dependent on the endogenous
nucleophiles including glutathione, methionine, metallothionein, and
other cellular components [2,25]. The endogenous nucleophiles act as a
defense mechanism to counter attack cisplatin induced toxicity at lower
levels of exposure.

Figure 5: Representative Comet assay images of HL-60 cells exposed to cisplatin 0, 1, 2, or 3 µM for 24, 48, 72, and 96 h. The treated and untreated
cells were subjected to single cell gel electrophoresis, stained with SBGR, and analyzed as described in the Materials and Methods section.
In this study, HL60 cells were treated with 1, 2, or 3 µM cisplatin for
various time periods. Study results show a significant cytotoxic effect
in all tested doses (Figure1). The lowest tested dose, 1 µM cisplatin,
inhibited almost 60% of cell proliferation compared to the controls. The
anti-proliferative or anti- cancer properties of cisplatin are in agreement
with the previous reports that cisplatin inhibited cell proliferation rate in
various cancers [26-28].
The eventual inhibition of cell proliferation or cytotoxicity in cells could
be the result of collective mechanisms of cisplatin-induced oxidative stress,
genotoxicity and other cellular responses. Hence, we determined whether
oxidative stress plays a key role in cisplatin-induced toxicity in HL60
cells. We found that the activities of antioxidant enzymes such as SOD
and catalase significantly increased in cisplatin-treated cells compared to
control cells. These increases in enzymatic activities were both dose- and
time-dependent (Figures 2 and 3).
As a part of defense mechanism, SOD catalyzes the dismutation of
superoxide anion to hydrogen peroxide (H2
O2
) and the H2
O2 become
the substrate for the catalase. The induction of these two enzymes in the
present study can be considered as an adaptive mechanism to combat
the reactive oxygen species induced by cisplatin exposure. The dose-and
time-dependent increases in antioxidant levels in the present study are
consistent with the previous reports on cisplatin-induced reactive oxygen
formation in a concentration- and time-dependent manner [15]. As a
result of elevated antioxidants in response to oxidative stress, we observed
a significant induction of MDA, alipid peroxidation byproduct, in HL60
cells treated with cisplatin (Figure 4). It has been previously reported
that the release lipid peroxidation products increases carbonylation of
proteins, induces oxidative damage of cell membranes, and those events
may lead to the initiation of cell death [29-33].
In addition to oxidative stress, cisplatin induces genotoxocity and it is
probably the most effective way to inhibit cell proliferation or cell cycle
progression. Reactive cisplatin has been reported to react with functional
residues such as sulfhydryl groups in proteins and DNA, specifically,
nucleophilic sites of purines in DNA, leading to the formation of DNAprotein
and DNA-DNA inter strand or intra strand cross links [34-37].
In the present study, the reported DNA damage was dose- and timedependent
(Figures 5 and 6). For 96 h treatment, DNA damage reported
for 1 µM is almost 5% whereas for 3 µM is almost 10% higher than the
respective controls (Figure 6). These findings are in agreement with
cisplatin induced oxidative stress (Figures 2 and 3) and lipid peroxidation
(Figure 4) in a dose-and time-dependent fashion. The induced DNA
damage could be result of inter and intra strand cross links or culmination
of both oxidative stress and DNA cross links. The current findings are
consistent with previous reports that cisplatin interacts with purine bases
of DNA and form DNA adducts to induce toxicity [8,38].
Cisplatin has also been reported to induce apoptosis and changes in
cell cycle progression in wild type p53 and p53 deleted hepatoma cell
lines [39]. The HL60 cells lack p53 protein, while cells with wild type
p53 have been reported to show more sensitivity to cisplatin compared
to p53-deficient cells [40]. These studies suggest that p53 activation
could sensitize the cells to cisplatin toxicity. It has been reported that
cisplatin induces apoptosis through both p53 dependent and independent
pathways [39,41,42]. In consistence with the results of our study, it has
been pointed out that cisplatin inhibits cell proliferation irreversibly by
arresting the cells in G1 phase of cell cycle [43]. However, additional
studies are required to elucidate the molecular mechanisms of cisplatininduced
growth inhibition, oxidative stress, cell cycle modulation, and
genotoxicity in cancer cells.
Conclusion
In this study, we investigated cisplatin-induced cytotoxicity, oxidative
stress and genotoxicity at low levels of exposure (1, 2, and 3 µM) and at
various time periods (24, 48, 72 and 96h). Cisplatin significantly inhibited
cell proliferation, and induced antioxidants levels and lipid peroxidation
even at lowest dose of 1 µM. Also, cisplatin significantly induced DNA
damage in HL60 cells at all treatment doses. Taken together, low doses of
cisplatin show a significant activity against HL-60 cells, and hence, may
be used in combination with arsenic trioxide (ATO) to improve treatment
and reduce potential side effects in acute promyelocytic leukemia patients.
However, additional research is required to study the combined effect of
cisplatin and ATO in order to determine their potential for use in APL
chemotherapy.
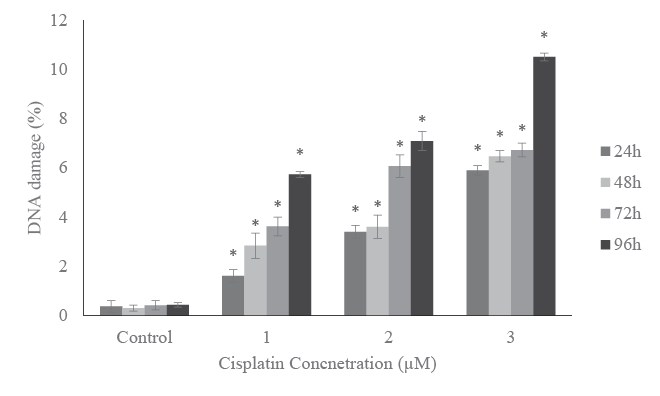
Figure 6: DNA damage in HL-60 cells treated with 0, 1, 2, or 3 µM
cisplatin for 24, 48, 72, and 96 h. Data represent the percentages of
DNA damageexpressed as means of three independent experiments
± standard deviations. p-values less than 0.05 were considered
statistically significant.
Acknowledgements
The research described in this publication was made possible in part by
a grant from the National Institutes of Health (NIMHD-G12MD007581)
through the RCMI-Center for Environmental Health at Jackson State
University, and in part by a grant from the Mississippi INBRE (NIGMSP20GM103476).