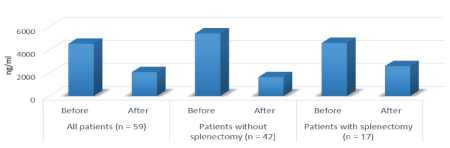
Figure 1: Mean Ferritin before and after thalidomide treatment
Vijay Ramanan* Ketki Kelkar
Yashoda Hematology Clinic, Pune Mangalmurti Complex, 109, First floor, Near Hirabaug Chowk, Lokmanya Bal Gangadhar Tilak Road, Pune, Maharashtra, India*Corresponding author: Vijay Ramanan, Yashoda Hematology Clinic, Pune Mangalmurti Complex, 109, First floor, Near Hirabaug Chowk, Lokmanya Bal Gangadhar Tilak Road, Pune, Maharashtra, India, E-mail: bmtpune@gmail.com
Objective: To assess the effect of thalidomide on ferritin levels and duration of blood transfusion in patients with β-thalassemia.
Background: Thalassemia is the result of defective hemoglobin production due to reduced or absent expression of β- globin gene. Currently, the main therapy in β-thalassemia patients is a regular blood transfusion schedule and use of iron chelating agents. However, they are associated with limitations and severe therapeutic complications. Thalidomide can reduce α-globin chain production in erythroid progenitors and improve α:β chain imbalance. Few case reports indicate that thalidomide should be considered in cases of thalassemia which cannot be treated with transfusions and do not respond to hydroxyurea.
Design: Retrospective study
Subjects and method: Medical records of thalassemia patients who received thalidomide between January 2006 and April 2016 in our institution were reviewed. Data gathered included age, sex, splenectomy status, ferritin levels before and after treatment, duration of off blood transfusion, first blood transfusion was analyzed.
Results: the study included 104 subjects (34 females and 70 male) with an age ranging from 1 to 36 years (mean age: 13.04 years). Out of this, 27 patients had undergone splenectomy. Data of ferritin levels before and after treatment were available for 59 patients. Ferritin levels reduced to 51% in all patients (4037 to 2086 ng/ml). Ferritin levels reduced to 55% in patients who had undergone splenectomy and 49% in patients who had not undergone splenectomy. Age at which first blood transfusion started ranged from 3 months to 7.5 years (mean: 16.6 months). Duration of off blood transfusion ranged from 1 month to 42 months (mean: 10.22 months). There were 9 cases with βº- Thal Homozygotes and 4 cases of β+-Thal Homozygotes or β+/βº Compound Heterozygotes.
Conclusion: Thalidomide treatment reduced ferritin levels. Further studies are required to define the potential use of thalidomide in thalassaemia.
Thalidomide; Beta thalassemia; Myeloma treatment
Thalassemia is a well-known genetic disorder of blood characterized by reduced or lack of the globin chain synthesis. Thalassemia is a condition in which person is unable to produce normal oxygen carrying components [1]. Thalassemia was first reported in Mediterranean population. This condition is more pronounced in thalassemia belt region of the world. Around 15 million population is suffering from this disorder and about 240 million population are reported as the carrier. In India alone the β-thalassemia carrier varies from 1 to 17% (average of 3.2%) i.e. 1 in every 25 people in India are the carrier of thalassemia [2,3]. Thalassemia is common in malaria endemic region, as an abnormal RBC does not support malaria parasite to complete it life cycle.
Beta thalassemia is a type of thalassemia, in which Beta globin chain is compromised. Excess formation of α-chain initiates during erythropoiesis and reaches maximum concentration at polychromatophilic erythroblasts, results in apoptosis of cell [4].
Thalassemia has limited treatment option. Management of thalassemia is channeled via treatment of severe anemia, prevention of undue erythropoiesis and iron overloading [5,6]. In severe form of anemia, blood transfusion is the mainstay at delineate frequency. Although few patients developed unmanageable antibodies result to untransfusable and iron overloading casing dame to vital organs. Therefore, chelation therapy is obligatory in those cases along with single or in combination. Renal tubular damage, including Fanconi’s Syndrome, has been reported in patients treated with deferasirox (one of the iron chelator), most commonly in children and adolescents with beta-thalassemia and serum ferritin levels <1500 mcg/L [7].
Though various options for management of thalassemia available currently, yet there are limitations with the existing therapies, hence there is an unmet need for newer agents. Thalidomide, an old drug used for multiple myeloma treatment has appropriate effects in HbF induction with similar mechanism of action with other immunomodulatory agents. The exact mechanism of the therapeutic effect of this factor is still unknown. However, the effects of thalidomide may be due to suppression of NFKB induction by inflammatory cytokines such as Tumor Necrosis Factor (TNF-α), Vascular Endothelial Growth Factor (VEGF) and prostaglandin E2 synthesis (PG-E2) associated with increased release of reactive oxygen species (ROS). ROS can launch P38 MAPK, which results in increased HbF levels. The major side effects of thalidomide were reported as somnolence, Constipation, Gynaecomastia, DVT. Adult patients were advised to avoid pregnancy during treatment [8].
This paper will discuss about the thalidomide use in patients with different medical condition.
We conducted a retrospective study to analyze the outcome of treatment with thalidomide in thalassemia patients. We recruited thalassemia patients, who received treatment with thalidomide between 1-Jan-06 and 21-Apr-16 in our institute. Thalidomide was used in reduced dose (2 mg/ kg to 10 mg/kg) which was derived from another in house study in patients with graft rejection. The following parameters were recorded: age and sex, hemoglobin (Hb) levels, status of splenectomy, ferritin levels before and after treatment with thalidomide, age at which first blood transfusion was given first and duration in months without blood transfusion (off BT). Thalassemia mutation analysis and Hb electrophoresis data were analyzed in some patients.
One hundred and four subjects (34 female and 70 male) were included in the study (Table 1). The mean age of the patient was 13.04 years (range 1-36 years; standard deviation (SD) ± 8.39 years). Twenty-seven individual had under gone splenectomy (20 men and 7 women) prior to treatment. Values of ferritin levels for before and after thalidomide treatment were available for all 104 patients; data of only these patients was analyzed. Ferritin levels before thalidomide treatment ranged from 987.5 to 13,290 ng/mL (mean: 4534 mg/mL). Mean duration of thalidomide therapy was 14 months (6 to 24 months).
| Total number of patients | 104 |
| Age (mean) in years of the whole study population | 1 to 36 (13) |
| Number of Males | 70 |
| Number of Thalassemia Major (TM) patients | 102 |
| Number of Thalassemia Intermediate (TI) patients | 1 |
| Number of Thalassemia carrier patients | 1 |
| Age (mean) in years | 1 to 36 (13.6) |
| Female | 34 |
| Age (mean) in years | 1 to 36 (13) |
| Hemoglobin range (mean) | 4.8 to13 (8.71) |
| Splenectomy | 27 |
| in male | 20 |
| In female | 7 |
Table 1: Profile of patients enrolled in the study
Ferritin levels after thalidomide treatment ranged from 100 to 11,000 ng/mL (mean: 2061.15 ng/mL).Thalidomide treatment significantly was reduced the ferritin levels (p<0.00001, paired t-test). Seventy seven patients had not undergone splenectomy; mean ferritin levels before and after thalidomide treatment were 5756 and 1847 ng/mL in these patients. The reduction in ferritin level following thalidomide treatment was significant in those who did not undergo splenectomy (p<0.00001). There were 42 male patients, who had not undergone splenectomy; mean ferritin levels in these patients before and after thalidomide was 5123 and 1446 ng/mL. Thirty five women had not undergone splenectomy; mean ferritin levels before and after thalidomide treatment were 3195 and 1963 ng/mL. There were 27 patients, who had undergone splenectomy; mean ferritin levels before and after thalidomide treatment was 4601 and 2588 ng/mL in these patients. The reduction in ferritin level following thalidomide treatment was also significant in those who underwent splenectomy (p=0.0031) (Figure 1).
Figure 1: Mean Ferritin before and after thalidomide treatment
Fourteen patients of age 2.5 years to 27 years were observed for the increase in HbF level according to their age. Thirteen subjects were found with HbF level 75% to 101.4 % in comparison to normal HbF level in patients without thalassemia. Only one subject was identified to have thalassemia carrier (i.e. HbF is about 6.3%) (Figure 2). Maximum patients had mutations of IVS1-5(G-C), IVS1-1 (G-T), and 619 bp in that order. Data of age at which first blood transfusion was available for 82 patients and it ranged from 3 months to 7.5 years (mean: 16.6 months). Ninety-seven patients were off blood transfusion for a duration ranging from 1 month to 42 month (mean: 10.22 months). In patients who had undergone splenectomy, mean duration of off blood transfusion was 10 months. In patients who had not undergone splenectomy, mean duration of off blood transfusion was 9.9 months. Frequency of blood transfusion was in between 10 days to 45 days.
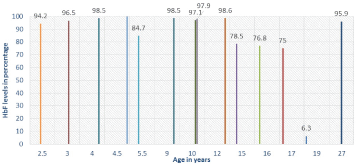
Figure 2: HbF level with respect to age of patients
Out of total 104 patients, 2 cases of DVT and 2 cases of Gynecomastia were observed. Patients with DVT were discontinued on Thalidomide therapy and started on Hydroxyurea therapy.
Thalassemia is characterized by defective production of the β-globin molecule [1]. The conventional treatment for these patients includes regular blood transfusions and chelating therapy. Fetal hemoglobin inducers are a promising therapeutic strategy for patients with beta thalassemia major and other hemoglobinopathies. Higher production of γ-globin lowers the imbalance between β and non-β-chains and thus reduces hemolysis. Consequently, by improving the ineffective erythropoiesis, hemolysis can be alleviated, and owing to improved survival of red cells containing higher levels of fetal hemoglobin (HBF), the total hemoglobin levels can be increased. Hydroxyurea and histone deacetylase inhibitors have been studied as HBF inducers.2 Thalidomide is found to induce β-globin gene expression and increase the proliferation of erythroid cells in in vitro culture models [2,9-11].
Except for few case reports, there is no data on the treatment effect of thalidomide in thalassemia patients. We are the presenting a retrospective data on thalassemia patient, who were treated with thalidomide. In our study, 25.9% of patients had undergone splenectomy. Ferritin levels reduced to 46.5 % of the initial value in all patients (4534 to 2061 ng/mL; 54.5% reduction). In patients, who had undergone splenectomy, ferritin levels reduced to 61% (reduction by 39%) and in patients who had not undergone splenectomy, it reduced to 32% (reduction by 68%).
Three case reports, reported that thalidomide therapy at 50 to 100 mg/ kg per day caused a progressive and rapid increase in total hemoglobin and HBF levels in β-thalassemia major patients [12,3].
Lopez, et al. [13] reported a case study of a 21-year-old women with β thalassemia major diagnosed at 5 months. Her hemoglobin levels without transfusion were as low as 2.9 g/dL. She had chronic blood transfusions, every 2 or 3 months, with an iron overload. She was splenectomized at the age of five years. She had received chelation therapy (Desferoxamina) with different time intervals. She was treated with thalidomide (100 mg per day). Hemoglobin increased after three months to 7 g/dL. Thalidomide was given continuously and never was transfused again with hemoglobin levels stabilizing between 7.6 and 10.6 g/dL and achieving almost 100% HBF [12].
Masera N, et al. [14] reported a case of a young girl with β-thalassemia (β+/β) in a severe condition, who could not be given any further transfusions because of severe post-transfusion reactions. From the age of 1 year, she was transfused every 3-4 months, maintaining very low hemoglobin values (in the range of 5-7 g/dL). Splenectomy was performed when she was 4 years old. When she was 9 years, repeated transfusion attempts proved ineffective. At 10 years, she was started on hydroxyurea (10 mg/kg/day) and there was a partial response. At 15 years of age she was treated with three cycles of rituximab, and no response was seen in terms of hemoglobin levels. She was stable with hydroxyurea for some time but a progressive decrease in hemoglobin was observed. There were no signs of infection but she was clinically worsening with severe heart failure and initial lung edema. She was started on treatment with thalidomide 75 mg/ kg/day and her Hb progressively and rapidly increased. One month after starting thalidomide treatment, Hb was 7.2 g/dL; after 8 months: Hb was 9.0 g/dL, HBF: 73%. The levels of erythroblasts remained high, although showing a slight decrease (53 × 103 /mL). The response to thalidomide was good and she tolerated the treatment well [15].
A recent study reported two cases in which thalidomide was used as erythroid stimulating agent for non-transfusion dependent thalassemia (NTDT). A 48-year-old woman had been diagnosed with NTDT at the age of 6 years. Splenectomy was performed at the age of 32. At the age of 42 years, she required chronic transfusion treatment. Soon she developed severe post-transfusional alloimmune hemolytic anemia [16]. High dose steroids plus azathioprine 100 mg and rituximab 375 mg/m2 up to eight cycles also failed to increase Hb. Thalidomide 50 mg/100 mg on alternate days increased Hb level to 9.99 g/dL within the first month, a dose reduction for thalidomide to 50 mg daily also maintained Hb at 10.7 g/dL. Thalidomide dose was further reduced to 50 mg every other day and was stopped with a Hb of 10 g/dL. Within one month Hb dropped to 8.75 g/dL but, after restarting thalidomide at a dose of 50 mg for 5 days a week, a further response was achieved within one month. Ten months after starting treatment with thalidomide, the patient had responded with Hb above 10 g/dL. In the second case, the patient had presented with homozygosity for the βº 39 mutation, and r α-thalassemia, and she had received her first transfusion at the age of 13 years and was splenectomized at the age of 28 years. At the age of 48, she developed a recurrent post-transfusional alloimmune hemolytic anemia. After high dose steroid failed, she was started on thalidomide 50 mg/100 mg on alternate days together with mycophenolate mofetil 500 mg/day and high dose poly-specific IV Ig (5 cycles with 20 g for 5 days up to day +115 of thalidomide therapy). Her Hb level rose to 3.3 g/dL within the first month and to 6.5 g/dL in the second month. Thalidomide and mycophenolate doses were slowly tapered. Her Hb remained above 8 g/dL (HbF=98%) after 4 years of continuous treatment with thalidomide, with no detectable thalidomide-related side effects [3].
Fetal hemoglobin induction is a promising treatment option for β-thalassemia and sickle cell disease as it can improve clinical symptoms of these genetic disorders. Thalidomide has made a remarkable comeback since the discovery of its immunomodulatory and anti-inflammatory effects, which have led to its use as a treatment for various proinflammatory and autoimmune conditions. Thalidomide has been used in few severe thalassemia cases, after failure of various immunomodulatory approaches. Thalidomide should be considered in cases of thalassemia. Further biological and clinical studies are required to define the potential use of thalidomide in thalassemia and other hemoglobinopathies.
Download Provisional PDF Here
Article Type: Research Article
Citation: Ramanan V, Kelkar K (2017) Role of Thalidomide in Treatment of Beta Thalassemia. J Blood Disord Med 3(1): doi http://dx.doi.org/10.16966/2471-5026.119
Copyright: © 2017 Ramanan V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access