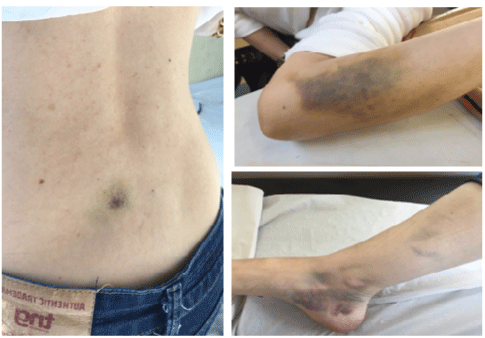
Figure 1: Spontaneous bruising on the back, left forearm and rigth lower limb
Daniela de Oliveira Werneck Rodrigues1* Irtis de Oliveira Fernandes Junior2 Adriana Aparecida Ferreira1 Lysla Cardoso Sudário2 Bruno Almeida Rocha Maciel2 Dandara Emery Morais Sana2 Nathalia Chebli de Abreu2 Maisa Marques Magalhães2
1Fundação Hemominas, Brazil*Corresponding author: Daniela de Oliveira Werneck Rodrigues, Hemominas Juiz de Fora Foundation MG Rua Barão de Cataguases, Juiz de Fora-MG, Brazil, Tel: +55 32 999796484 3126; Fax: 32 3257 3100; E-mail: danielawerneckhemato@hotmail.com; daniela.werneck@hemominas.mg.gov.br
Acquired Hemophilia A is a rare entity, resulting from the production of autoantibodies against factor VIII of the coagulation process. The presence of these autoantibodies, usually idiopathic, may be related to autoimmune conditions, drugs, neoplastic diseases and pregnancy. The diagnosis involves clinical aspects and laboratory findings, such as prolonged activated partial thromboplastin time, decreased levels of factor VIII and the presence of autoantibodies against it. The clinical manifestations of Acquired Hemophilia A are characterized by mucous-cutaneous, intramuscular, and/or postpartum bleeding. The treatment for Acquired Hemophilia A includes immunosuppressant drugs, reducing hemorrhages and intervening on the etiology of the inhibitor’s production. The authors report a case of Acquired Hemophilia A associated with pregnancy with several muco-cutaneous hemorrhagic manifestations and intramuscular hematomas with excellent response to the use of immunosuppressants.
Hemophilia A; Acquired hemophilia A; Immunosuppression; Inhibitor; Factor VIII
The most frequent causes of post-partum bleeding (more than 500 ml of blood through the vagina in 24 hours) are related to uterine atony (68% of cases), placental retention (28.3%) and perineal and birth canal lesion (3%) [1-3]. Coagulation disorders are responsible for 0.5% of the causes of bleeding during the puerperium and should be taken into account whenever the parturient does not respond to uterine atony treatment (uterotonics, uterine tamponade, clamping or arterial embolism), and there is no placental remnants and signs of birth canal lesion [4-6].
The hemostasis disorders related to postpartum bleeding are: HELLP syndrome, Von Wille brand disease, coagulation factors deficiency (VII, VIII, XI, XIII and fibrinogen), Glanzmann thrombasthenia, BernardSoulier syndrome, gray platelets syndrome, Acquired Hemophilia A (AHA) and others [2,7]. AHA is a rare autoimmune entity, marked by the presence of autoantibody against factor VIII of coagulation [8-11].
AHA affects 1.2 to 1.4 per million people a year, being more common in men over 60. As well as in the hereditary form of haemophilia, AHA can occur in all ethnic groups and has a worldwide prevalence. Acquired hemophilia typically presents in middle age and beyond and rarely arises in childhood [12]. In the 20-30 age groups, it is more prevalent among women, because of the relationship with pregnancy and the puerperal period, with a frequency of 7-21%. During pregnancy and puerperium, AHA can manifest through cutaneous, vaginal and retroperitoneal bleeding [13-18].
The possible etiologies for AHA are described in (Table 1). Regarding malignant neoplasm, lung and intestine tumors are the most causes related to AHA development [15,16].
| Pregnancy and post- partum | Allergic reactions on drug | Solid Tumors |
| Autoimmune diseases | penicillin’s and derivatives | Prostate |
| Systemic lupus erythematosus | Sulfamides and quinolones | Lung |
| Rheumatoid arthritis | Griseofulvin | Colon |
| Multiple sclerosis | Phenytoin | Pancreas |
| Temporal Arthritis | Chloramphenicol | Stomach |
| Sjögren's Syndrome | Methyldopa | Bile ducts |
| Autoimmune Hemolytic Anemia | Levodopa | Uterine cervix |
| Good Pasture syndrome | Interferon alpha | Head and neck |
| BCG vaccine | Breast | |
| Myasthenia Graves | Melanoma | |
| Graves's disease | Clopidogrel | Kidney |
| Autoimmune hypothyroidism | Antidepressants | Respiratory disease |
| Inflammatory Bowel Disease |
Hydralazine | Asthma |
| Idiopathic thrombocytopenic purpura | Acetaminophen | COPD |
| Hematological diseases | Diabetes | Dermatological diseases |
| Waldestron's disease | Acute hepatitis virus infection | Psoriasis |
| Leukemia | B Hepatitis | Pemphigus |
| LLC | C Hepatitis | |
| Hodgkin disease | H Hepatitis | |
| Myelofibrosis | ||
| Multiple myeloma | ||
| Myelodisplasic syndrome | ||
| Source: Mingot (2017) with permission | ||
Table 1: AHA causes
AHA is diagnosed through the presence of bleeding and the increase of activated partial thromboplastin time, without changes in platelet count, prothrombin time and thrombin time. The plasma level of factor VIII is low and there are inhibitors against this factor. The presence of the inhibitor is generally confirmed using a blood clotting assay called the Bethesda Inhibitor Assay. Antibody levels can be performed using this test and is described as the number of Bethesda units (UB). The exclusion of positive results of lupus anticoagulant is recommended, considering the possibility of cross reactions and false-positives that might interfere with laboratory analysis of inhibitors against factor VIII [19-21].
The investigation of drug use should be judicious regarding the use of anticoagulants such as heparin and antiplatelet agents (acetylsalicylic acid and clopidogrel) that may compromise the evaluation of hemostasis [15,22].
The treatment for AHA aims to remove the triggering factor, control bleeding and eradicate the inhibitors. Bleedings can be controlled with the use of coagulation factors, such as activated prothrombin complex concentrate and recombinant activated factor VII. The risk of thromboembolic events (heart attack, venous thromboembolism, and strokes) should be considered when activated coagulation factors are used [12,13,15,23].
Tranexamic acid, which is an inhibitor of fibrinolysis, can be used alone or associated with rFVIIa in bleeding control [13]. Inhibition and control of antibodies against FVIII is done with the prescription of immunosuppressant’s, with corticosteroids being the drug of choice. The recommended dose of prednisone is 1 mg/kg/day. This treatment is effective in most of the cases and should be maintained until the total disappearance of the antibodies against FVIII [22-24].
Corticotherapy can be used combined with other cytotoxic agents, such as cyclophosphamide, azathioprine, cyclosporine, vincristine, mycophenolate, and others. Currently, the use of rituximab, anti-CD20 monoclonal antibody, has been described in the drug approach of AHA. Rituximab is a recognized therapy for the treatment of proliferative hematological and autoimmune diseases [22-25].
Women who develop postpartum AHA may present spontaneous resolution of the condition, however immunosuppressive therapy substantially reduces the time to progress and the number of hemorrhagic complications associated with AHA.
Patients should be followed for up to one year after remission of hemophilia considering the risk of recurrence [26,27].
The aim of this case report on acquired postpartum haemophilia is to draw attention to a disease that, although rare, should be considered in case of unusual postpartum bleeding due to the possibility of complications and even death [12,28].
A 34-years-old Caucasian female, in your first pregnancy, underwent cesarean delivery in April of 2015 without complications. Forty days later she developed spontaneous hematomas manifested on legs with progressive worsening, besides intramuscular bleedings on the back, forearm, peri-malleolar region and thighs.
Her past medical history included an oophorectomy and a breast implant, without bleeding complications. She had no personal or family history of the hematologic or autoimmune disease. The physical examination found bruises on the right medial peri-malleolar region, on the left forearm, and on the back (Figure 1). The obstetrician service referred her to the hematological investigation.
Figure 1: Spontaneous bruising on the back, left forearm and rigth lower limb
The patient was admitted to the coagulopathy service at Fundação Hemominas in May of 2015. The laboratory screening tests for coagulation disorders showed an increase of aTTP by its own (Table 2). A plasma dosage of factor VIII, factor IX and Von-Willebrand factor was performed, followed by an examination for the presence of inhibitors against factor VIII. With the reduction of factor VIII levels (3.5%) and the presence of inhibitors against it (Inhibitors title:10UI Bethesda), the possibility of AHA was considered. The diagnosis of Von Willebrand disease or platelet disorders was disregarded. Research on the presence of lupus anticoagulant, antiphospholipid antibody syndrome, autoimmune diseases (anti-nuclear factor, Anti-DNA, anti-SM, anti-RNP and rheumatoid factor), infectious diseases (human immunodeficiency virus, C and B hepatitis, syphilis), research on paroxysmal nocturnal hemoglobinuria (CD55/CD59/Flair), screening for neoplasms (carcinogenic antigen, CA 19.9, CA 15.3, CA 125 and lactate dehydrogenase) were negative.
| Initial Exams | Normal Values | |
| Activity of Prothrombin | 13s (13) 100% | 100% |
| Fibrinogen | 430 mg/dL | 175-450 mg/dL |
| Partial Thromboplastin time | 54s (28) | 28-38s |
| actived (a PTT) | ||
| Test for diagnosis | ||
| aPTT | 62s (28) | 28-38s |
| FVIII | 3,50% | 50-150% |
| FIX | 124% | 50-130% |
| Inhibitor of factor VIII | 10 UB* | Negative 0 UB |
| Low title<5 UB | ||
| High title>5 UB | ||
| Source: authors | ||
| UB*: Bethesda Units |
Table 2: Test for diagnosis and investigation
The patient was treated with prednisone 1 mg/kg/d plus tranexamic acid and showed a gradual and total improvement. There was a normalization of plasma levels of Factor VIII, without the presence of inhibitors against FVIII (Table 3). The last control was performed in March of 2017 when the patient was asymptomatic (Figure 2).
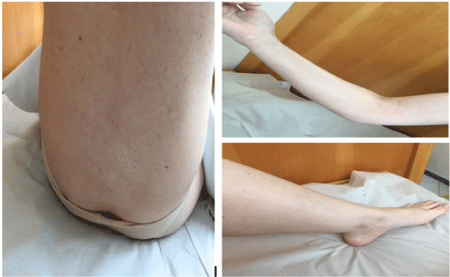
Figure 2: Resolution of the hemorrhage after corticotherapy
| jul/15 | aug/15 | oct/15 | dec/15 | jan/16 | |
| Inhibitor | 2.5 UB* | negative | negative | negative | negative |
| Factor VIII | 3.5% | 8.0% | 12% | 35% | 50% |
| aPTTa | 62.5 (30) | 46.5 (31) | 35.8 (27.3) | 34.8 (28.2) | 32 (28) |
| Source: authors | |||||
| *UB = Bethesda Units | |||||
Table 3: Follow-Up Tests
AHA is a rare coagulation disorder marked by a decrease in plasma FVIII levels due to the presence of an active inhibitor against it, manifested clinically through muco-cutaneous and intramuscular bleeding, which makes it different from congenital hemophilia A, in which hemarthrosis is more common [8,12,13,24,29,30].
Several etiologies are known to produce antibodies against FVIII such as pregnancy, drugs, autoimmune diseases and malignant neoplasms.
Treatment for AHA involves control of bleeding, elimination of the causative factor that precipitated the production of antibodies against factor VIII, and reduction of plasma levels of the inhibitor through immunosuppressive therapy [23,26,31].
AHA occurs rarely, but develops suddenly and occasionally presents with life-threatening bleeding, with a high mortality rate, estimated between 9 to 33% [23], it has decreased in tandem with the advancement of therapeutic interventions since the 1980s. The late morbidity and mortality are more associated with the secondary effects of the immunosuppressive drugs used for the eradication of the inhibitor [12,23].
The authors report a case of AHA with the positivity of inhibitors against factor VIII related to gestation/postpartum period. The clinical manifestation was through cutaneous and intramuscular bruises made evident 40 days postpartum, a period comprehended within the expected range, up to the second month of the puerperium, according to Poblet 2015 [12,23,32], Sebastian described et al. [18], a similar case of AHA during pregnancy, manifesting through multiple and large bruises on the legs. Other studies, as in Sheetala 2013 and Lee 2011, show distinct manifestations, such as excessive vaginal bleeding during the puerperium [17].
Mo and Bao presented two cases of severe AHA in Chinese women in 2017, one of these women developed this disorder in the setting of possible parvovirus B19 infection, and the other woman failed to respond to usual first-line therapies despite exhibiting a less severe clinical course, illustrating the varied but potentially stubborn behavior of this disorder [27].
In our case, there was no personal or family history of bleedings, which is expected and common in AHA, as described by Lee 2011. The examination for the neoplastic and autoimmune disease was negative. The woman was treated with prednisone 1 mg/kg/day, with excellent results [24,29,33].
Although rare, it is important to consider the diagnosis of AHA for cases of puerperal bleeding in which the treatment based on its most common causes (uterine atony, the presence of placental remnants or birth canal lesion) is unsatisfactory. To improve the forecast, an early diagnosis that facilitates appropriate treatment in time is fundamental. It is essential to disseminate knowledge about this entity among the global medical professionals.
To Dr. Maria Eva Mingot-Castellanos for the contribution.
The authors declare that there is no conflict of interests regarding the publication of this paper.
Download Provisional PDF Here
Article Type: Case Report
Citation: Rodrigues DO, Junior I, Ferreira AA, Sudário LC, Rocha Maciel BA, et al. (2017) Postpartum Acquired Hemophilia A: Case Report and Literature Review. J Blood Disord Med 2(1): doi http://dx.doi.org/10.16966/2471-5026.117
Copyright: © 2017 Rodrigues DO, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access