Introduction
Down observed in 1866 that ‘Those who have any given attention to
congenital mental lesions, must have been frequently puzzled how to
arrange, in any satisfactory way, the different classes of this defect which
may have come under their observation. Nor will the difficulty be lessened
by an appeal to what has been written on the subject. The systems of
classification are generally so vague and artificial, that, not only do they
assist but feebly, in any mental arrange of the phenomenon which are
presented, but they completely fail in exerting any practical influence on
the subject’ [1].
Such frustration applies just as fittingly to the syndrome of
microangiopathic hemolytic anemia (MAHA) and thrombocytopenia.
Patients presenting with the syndrome of MAHA and thrombocytopenia
(MAHA/T) have been given the diagnosis of thrombotic thrombocytopenic
purpura (TTP), hemolytic uremic syndrome (HUS), TTP/HUS or TTP/
TMA (thrombotic microangiopathy). Almost invariably the criteria for
these diagnostic terms are vague, artificial or both.
A serious consequence of indiscriminate usage of diagnostic terms is
that all patients presenting with the syndrome of MAHA/T are treated as
one disease, ‘TTP’, with plasma exchange, supplemented with anti-platelet
agents, corticosteroids, immunosuppressive drugs, and even splenectomy.
On the other hand, the correct diagnosis is missed for patients who do not
have MAHA, thrombocytopenia or both.
Difference between Syndromes and Diseases
Many diseases are first recognized as a syndrome, which is simply a
particular collection of symptoms and/or signs. It is intuitively assumed
that a syndrome is the consequence of a particular disease etiology or
pathogenesis. When the etiology of the syndrome is identified, it becomes
a disease, although often under the same or similar term. The discovery
of the etiology allows exclusion of patients with different etiology etiology
or pathogenesis, and identification of patients with atypical features of
the disease. Almost invariably the disease is found to be more variable
in manifestations than previously recognized. Down syndrome took this
path after the discovery of trisomy 21 in 1959 as its cause [2].
Unfortunately, not all syndromes are due to one particular etiology or
pathogenesis. Defining ‘TTP’ or ‘TTP/HUS’ as a syndrome of MAHA
and thrombocytopenia is a conspicuous example of how syndrome-based
disease definition may go seriously astray. Until recently, ‘TTP’ or ‘TTP/
HUS’ has been a source of much confusion and uncertainty. Some have
tried to rectify the confusion by adding criteria such as young age and
severe renal dysfunction for atypical HUS, and fever and neurologic
deficits for TTP. Such conditions are artificial and do not resolve the
intrinsic uncertainty of epiphenomenon criteria.
From Syndromes of TTP or TTP/HUS to Diseases of STXHUS,
TTP, AHUS and Others
Among the patients presenting with the syndrome of MAHA/T,
a subset, first recognized in young children, have a prodrome of
hemorrhagic diarrhea and are often afflicted with prominent renal
failure [3]. This subset of patients, often given the diagnosis of typical or
diarrhea+ hemolytic uremic syndrome (D+HUS), are found at autopsy
to have thrombotic microangiopathy (TMA), which is characterized with
endothelial injury and thrombosis in the small arteries, arterioles and
glomerular capillaries in the kidney [4].
The hemorrhagic diarrhea of D+HUS was demonstrated in most
cases to result from colitis due to infection of shiga toxin producing E.
coli [5]. Subsequent analysis of stools for shiga toxins or shiga toxinproducing
E. coli shows that not all cases with the infection have obvious
hemorrhagic diarrhea before they go on to develop the complications of
TMA. Furthermore, some patients who develop TMA following such
infections do not have MAHA, thrombocytopenia or both; and the renal
insufficiency may be mild. To encompass these forme frusta cases, it is
necessary to replace the syndrome of D+HUS with the disease of shiga
toxin associated HUS (STX-HUS), which is TMA following infection with
shiga toxin-producing microorganisms, most commonly E. coli serotype
O157:H7. This etiologically defined disease includes most of the cases
previously given the diagnosis of typical or D+HUS, but also includes
the forme fruste cases that do not have MAHA, thrombocytopenia, or a
prodrome of hemorrhagic diarrhea.
In children, the syndrome of MAHA/T is also often associated with
prominent renal dysfunction even when it is not preceded by a prodrome
of hemorrhagic diarrhea or shiga toxin associated colitis. Such cases have
been given the diagnosis of atypical hemolytic uremic syndrome (AHUS).
On the other hand, most adult cases presenting with the syndrome of
MAHA/T do not have a hemorrhagic prodrome or evidence of stool shiga
toxins, and have normal or mildly abnormal renal dysfunction. These
adult patients are given the diagnosis of TTP. Many adult hematologists
believe, incorrectly, that AHUS only occurs in children and the small
numbers of adult cases that do develop severe renal dysfunction merely
have TTP with exceptionally severe renal injury.
In recent years, ADAMTS13, a circulating metalloprotease, was
identified and cloned during research to understand how the von
Willebrand factor (VWF) multimers in normal plasma are generated [6].
In the circulation, ADAMTS13 prevents the activation of VWF by cleaving
the large polymeric protein whenever its conformation is being unfolded
by shear stress. Deficiency of ADAMTS13, due to inhibitory antibodies
or genetic mutations, is found in essentially all patients who are given the
diagnosis of ‘TTP’ but do not have serious renal dysfunction (maximal
serum creatinine <2.5 mg/dl) or a potential cause such as pneumococcal
sepsis, pregnancy, autoimmunity with positive ANA tests, hematopoietic
stem cell therapy or chemotherapeutic drugs.
Separately, defective regulation of the alternative complement pathway
has been found in many children and adults given the diagnosis of ‘AHUS’
[7]. These findings show that ‘TTP’ with ADAMTS13 deficiency and
‘AHUS’ with defective regulation of the alternative complement pathway,
although similar in often presenting with MAHA and thrombocytopenia,
are indeed etiologically distinct.
Comparison of the pathological features of patients with ADAMTS13
deficiency and those with defective regulation of the alternative
complement pathway further reveals that these two disorders are also
quite different pathologically [8].
A review of the literature shows that clinicians have often used the term
of ‘TMA’ almost synonymously with the syndrome of MAHA/T. This
usage implies, incorrectly, that a patient presenting with MAHA/T have
endothelial injury (microangiopathy) and thrombosis in small vessels
and vice versa. On the other hand, pathologists often use the ‘TMA’ term
for any lesions with micro vascular thrombosis, even in patients without
MAHA and thrombocytopenia.
To avoid confusion, TMA should be used only for the pathological
syndrome of endothelial injury (microangiopathy) and thrombosis, as
is found in STX-HUS. AHUS associated with defective regulation of the
alternative complement pathway is also associated with the pathological
features of TMA. In ‘TTP’ with ADAMTS13 deficiency, the cardinal
feature of pathology is VWF-rich platelet thrombi in arterioles and
capillaries, accompanied with no evidence of endothelial injury. Thus,
TTP with ADAMTS13 deficiency.
The molecular tests developed in research have been translated into
clinical practice. Unsurprisingly, these tests identify patients that have
severe deficiency of ADAMTS13 activity (<10% of the activity in normal
plasma) or mutations affecting the regulation of alternative complement
pathway but do not have MAHA, thrombocytopenia, or both, although
many of these patients do have MAHA and thrombocytopenia on other
occasions. The tests also identify individuals with ADAMTS13 deficiency
or defective regulation of the alternative complement pathway but have
never been ill. These cases would be excluded from the conventional
syndrome-based diagnosis of ‘TTP’ or ‘AHUS’ until they present with the
complications later in life. However, this practice would be analogous to
excluding the diagnosis of sickle cell anemia for a patient who has the
hemoglobin βS
βS genotype but has not experienced a painful crisis.
Asymptomatic individuals found in family study to have genetic
deficiency of ADAMTS13 should be considered to have TTP because
the individuals are at risk of developing micro vascular thrombosis upon
exposure to stresses of infection, trauma, surgery or pregnancy. These
stresses may trigger intravascular VWF-platelet aggregation because they
may increase the release of VWF from endothelial cells, augment the
shear stress profile in the circulation, and/or further decrease the plasma
ADAMTS13 activity.
Similarly, asymptomatic individuals found in family study to have
defective regulation of the alternative complement pathway should be
considered to have AHUS because they are at risk of developing
TMA upon exposure to the stresses of infection, surgery, pregnancy or
intravenous radiographic contrast media. These stresses may trigger
uncontrolled complement activation, resulting in TMA with micro
vascular endothelial injury and thrombosis in the kidney.
Both groups of asymptomatic individuals likely have milder forms of
their diseases and thus require stronger triggers to precipitate intravascular
VWF-platelet aggregation in the case of ADAMTS13 deficiency or
uncontrolled complement activation and endothelial injury in the case
of defective alternative complement regulation. Diagnosis of TTP or
AHUS before complications occur provides the best opportunity to save the
individuals from serious and sometimes fatal consequences of the diseases.
To encompass such forme fruste cases, it is necessary to re-define
TTP and AHUS as etiology/pathogenesis-based diseases (Table 1). The
difference between mechanistically defined TTP and AHUS is further
delineated in table 2.

Table 1: Two different approaches to defining TTP and HUS
Abbreviations: ADAMTS13: Adisintegrin and Metalloprotease with thrombospondin type 1 repeats member 13; AHUS: Atypical hemolytic uremic syndrome; CFB: Complement factor B; CFH: Complement factor H; CFI: Complement factor I; D+: Diarrhea positive; D-: Diarrhea negative; ELISA: Enzyme-linked immunosorbent assay; HUS: Hemolytic uremic syndrome; MAHA/T: Microangiopathic hemolytic anemia and thrombocytopenia; STX: Shiga toxins; THBD: Thrombomodulin.
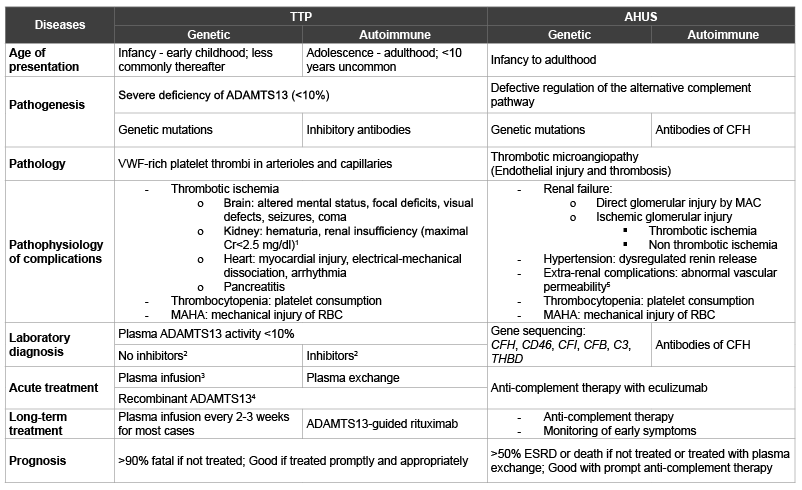
Table 2: Comparisons between TTP and AHUS
1Renal insufficiency can be more serious in genetic ADAMTS13 deficiency.
2ADAMTS13 inhibitors are only detected in 80%-90% of patients with the inhibitors. When inhibitors are not detected, onset during infancy or early
childhood and partial deficiency in first degree relatives favor genetic ADAMTS13 deficiency; Less-than-expected ADAMTS13 increase following plasma
exchange, increase of ADAMTS13 above 15% during remission and normal ADAMTS13 levels in first degree relatives favor autoimmune deficiency.
3Plasma exchange if the patient has renal failure with impaired urine output.
4Under development.
5Abbnormal vascular permeability may present as posterior reversible encephalopathy syndrome (PRES) of the brain; edema of brain, retina, bronchial
wall, alveoli, intestinal wall, mesentery, pancreas and/or cutaneous soft tissues; and fluids in pleural, pericardial and/or peritoneal cavities.
Abbreviations: ADAMTS13: Adisintegrin and Metalloprotease with thrombospondin type 1 repeat, member 13; AHUS: Atypical hemolytic uremic syndrome;
CFB, CFH, and CFI: complement factors B, H and I; ESRD: End stage renal disease; MAC: Membrane attack complex; THBD: Thrombomodulin; TTP:
Thrombotic thrombocytopenic purpura.
The complications of TTP include thrombocytopenia, due to platelet
consumption in thrombosis; MAHA, due to mechanical injury of the red
blood cells in the stenotic microvasculature; and ischemic dysfunctions
of organs such as brain, heart and kidney that are affected with micro
vascular thrombosis.
In contrast, the complications of AHUS include, in addition to
thrombocytopenia due to platelet consumption in thrombosis and
MAHA due to mechanical injury of red cells by abnormal levels of shear
stress, renal insufficiency, hypertension and extra-renal manifestations.
Renal failure in AHUS may result from direct glomerular injury by C5b9
(also known as membrane attack complex, MAC) or ischemic injury
due to microvascular stenosis, which may be caused by thrombosis,
non-thrombotic endothelial swelling and sub endothelial expansion, or
both. Hypertension results from dysregulated renin release due to preglomerular
hemodynamic disruption.
Extra-renal manifestations of AHUS such as headache, visual defects,
dyspnea, chest pain, abdominal pain, anorexia, nausea, vomiting, soft
tissue swelling and sudden death are associated with posterior reversible
encephalopathy syndrome (PRES) of the brain; edema of brain, retina,
bronchial wall, pulmonary alveoli, intestinal wall, mesentery, pancreas and/
or cutaneous soft tissues; and fluids in pleural, pericardial and/or
peritoneal cavities. These abnormalities are believed to result from
abnormal vascular permeability induced by anaphylatoxins C3a and
C5a of complement activation.
In AHUS, the pathology of TMA is found at autopsy primarily in
the kidney. It is assumed that the microenvironment of varying pH and
ionic strength in the kidney is conducive to complement activation;
anaphylatoxins C3a and C5a are likely released in the circulation from
the kidney. This may explain why extra-renal manifestations often abate
when a patient develops end stage renal disease and relapse after kidney
transplantation.
STX-HUS, TTP and AHUS are not the sole causes of MAHA and
thrombocytopenia. Overall, five different types of pathology have been
associated with MAHA and thrombocytopenia (Table 3). The pathological
lesions share the common feature of arteriolar stenosis, which generates
abnormal levels of shear stress and cause mechanical injury of the red
blood cells. Thrombocytopenia often accompanies MAHA because
thrombosis is the most common cause of arteriolar stenosis. In diseases
with non-thrombotic arteriolar stenosis, thrombocytopenia may occur
via other mechanisms, e.g. immune thrombocytopenia in vasculitis or
decreased megakaryopoiesis due to bone marrow metastasis in patients
with tumor cell embolism.
TTP and AHUS together account for the majority of cases presenting
with MAHA/T. In the author’s series, TTP and AHUS each account
for 60% and 20% respectively of the cases presenting with MAHA and
thrombocytopenia [9]. However, these figures are not intended to be taken
literally. Firstly, the case series includes many referrals whose diagnosis
the clinicians were uncertain. Secondly, the incidence of STX-HUS is
likely to vary widely, depending on geographic locations and occurrence
of outbreaks. The HELLP syndrome, the most cause of MAHA and
thrombocytopenia during pregnancy, may account for a larger fraction
of the cases encountered at institutions with busy obstetric services. The
incidence of MAHA/T also varies widely following hematopoietic stem
cell therapy [10,11].
Not included in table 3 are intravascular devices such as ventricular
assist devices, prosthetic heart valves and extracorporeal membrane
oxygenators that are also commonly associated with hemolysis due to
mechanical injury of red blood cells.
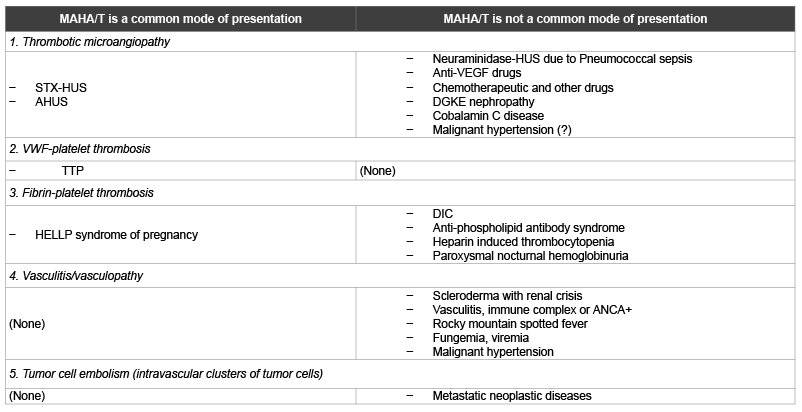
Table 3: Classification of MAHA/T based on pathological features in arterioles and capillaries
Abbreviations: AHUS: Atypical hemolytic uremic syndrome; ANCA: Anti-neutrophil cytoplasmic antibody; DGKE: Diacylglycerol kinase epsilon; DIC:
Disseminated intravascular coagulopathy; HELLP: Hemolysis, elevated liver enzymes and low platelet count; MAHA/T: Microangiopathic hemolytic anemia
and thrombocytopenia; VEGF: Vascular endothelial growth factor.
How the New Scheme affects the Diagnosis of TTP and
AHUS
Thrombotic thrombocytopenic purpura
Most cases of TTP are diagnosed when a patient presents with
thrombocytopenia and MAHA, especially at its very first presentation.
Neurological abnormalities such as altered mental status or focal deficits
are common but not invariably present. Fever occurs when the disease
is advanced. The kidney function is normal or only mildly impaired in
most cases. Nevertheless, advanced renal failure does not exclude TTP
because it may result from a concurrent disorder such as STX-HUS
[12], complement factor H mutation [13], or anti-glomerular basement
membrane nephropathy (a personal unpublished case).
Since most TTP patients are closely monitored after they achieve
remission, increasing numbers of relapses of TTP are diagnosed at the
stage of thrombocytopenia, before MAHA ensues. Occasionally, TTP
may present de novo as ‘idiopathic thrombocytopenia’, without MAHA.
TTP should be suspected when a patient who appears to be a case of
‘idiopathic thrombocytopenic purpura (ITP)’ also has symptoms such
as fatigue, headache or dizziness that is not otherwise explainable (Table
4). TTP should also be suspected when a patient presenting with stroke
or myocardial infarction has history of TTP or a decrease of the platelet
count from baseline levels that cannot be attributed to other causes.
Uncommonly, smoldering TTP may present with thrombocytosis due to
compensatory thrombocytopoiesis.
Confirming the diagnosis of TTP
When TTP is suspected, the diagnosis can be confirmed by ADAMTS13
analysis. The analysis includes plasma ADAMTS13 activity and inhibitors
of ADAMTS13. With a reliable assay, the plasma ADAMTS13 activity is ≤
10% in patients who have active platelet consumption. On the other hand,
a patient can be in clinical remission with ADAMTS13 ≤ 10%. When
ADAMTS13 activity is greater than 10%, VWF-platelet aggregation and
micro vascular thrombosis do not occur. A plasma ADAMTS13 activity
>10% during periods of persisting thrombocytopenia or declining platelet
counts excludes TTP as the cause of the thrombocytopenia or declining
platelet counts.
A plasma ADAMTS13 activity >10% but <20% (based on the mean
level of cases of MAHA and thrombocytopenia due to other causes, minus
3 standard deviations) is also likely to signify TTP. Such levels are most
commonly observed in TTP patient who have received transfusion of
blood products before the testing, but also occasionally in TTP patients
undergoing spontaneous remission.
Detection of ADAMTS13 inhibitors support the diagnosis of acquired
TTP due to autoimmunity of ADAMTS13. However, since the sensitivity
of assay is in the range of 80%-90%, a negative inhibitor assay result does
not exclude the diagnosis of autoimmune TTP. When inhibitors are not
detected, further investigation is needed to distinguish between hereditary
and acquired TTP. Findings that favor the diagnosis of hereditary TTP
include age of initial presentation less than 5 years and partial deficiency
of ADAMTS13 in first degree relatives. Conversely, plasma ADAMTS13
levels that increase less than expected after plasma therapy, or recover to
greater than 15% during remission, favor the diagnosis of acquired TTP
due to autoimmunity of ADAMTS13.
Plasma ADAMTS13 activity may be decreased in a variety of
pathological conditions such as sepsis, DIC, cirrhosis and multi organ
failures. The plasma ADAMTS13 activity level also decreases during
pregnancy, especially when it is complicated with the HELLP (hemolysis,
elevated liver enzymes and low platelet counts) syndrome [14,15]. However,
the plasma ADAMTS13 level in these pathological and physiological
conditions does not decrease below 10% to cause microvascular VWFplatelet
thrombosis. It should be noted that the plasma ADAMTS13
activity level may be 10% or lower, artifactually, if the blood sample is
properly handled and processed.
Atypical hemolytic uremic syndrome
Most cases of AHUS are suspected when the patients present with the
triad of renal insufficiency, MAHA and thrombocytopenia. The renal
insufficiency is often severe; yet it can also be mild, especially initially. Thus
the severity of renal function impairment does not reliably distinguish
AHUS from TTP. Other conditions that should raise the possibility of
AHUS are listed in table 4.
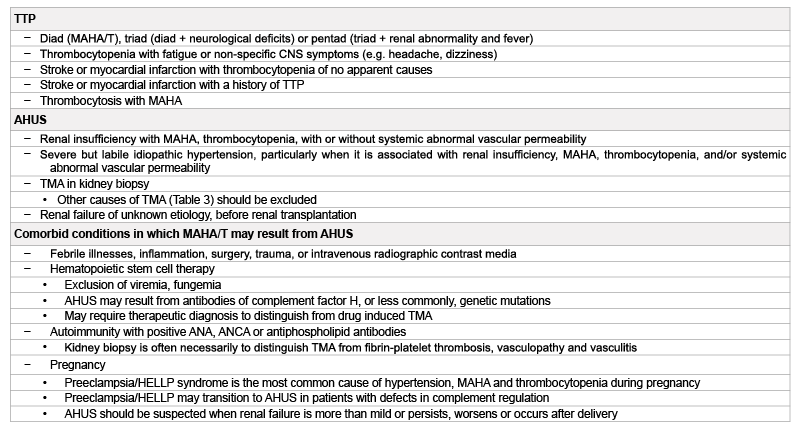
Table 4: Conditions in which TTP or AHUS should be suspected
Abbreviations: ANA: anti-nuclear antibodies; ANCA: Anti-neutrophil cytoplasmic antibodies; CNS: Central nervous system; HELLP: Hemolysis, elevated
liver enzymes and low platelet counts; MAHA/T: Microangiopathic hemolytic anemia and thrombocytopenia; TMA: Thrombotic microangiopathy; STX-HUS:
Shiga toxin associated hemolytic uremic syndrome; TTP: thrombotic thrombocytopenic purpura; VEGF: vascular endothelial growth factor.
In patients without severe ADAMTS13 deficiency and comorbid
conditions, AHUS is the presumptive diagnosis for those presenting
with renal insufficiency of any severity accompanied with MAHA,
thrombocytopenia and/or systemic abnormal vascular permeability.
TMA in kidney biopsy further supports the diagnosis.
However, AHUS is not the only cause of TMA. Other causes of
TMA should be excluded. These include STX-HUS, neuraminidaseHUS
in association with pneumococcal or other sepsis, anti-vascular
endothelial growth factor drugs, chemotherapeutic drugs, DGKE (diacyl
glycerol kinase epsilon) nephropathy and cobalamin C disease (Table
3). Nephropathy due to mutations of DGKE gene and, rarely, cobalamin
C disease may present as idiopathic TMA [16-18]. DGKE nephropathy
typically presents during infancy and its diagnosis can be confirmed by
mutation analysis of DGKE. Cobalamin C disease, due to mutation of
MMACHC (methylmalonic aciduria cblC type with homocystinuria) gene,
is characterized by elevated serum homocysteine and methylcobalamine
levels but normal folate and B12 levels [19]. Only rarely does it present as
idiopathic TMA.
Severe or malignant hypertension is often believed to cause the
syndrome of MAHA and thrombocytopenia and the pathology of TMA
[20-22]. Nevertheless, some of the cases are found in retrospect to have
AHUS [7]. AHUS may even present as severe or malignant hypertension
without MAHA, thrombocytopenia or renal failure [23]. AHUS
should be suspected in a case of hypertension that is severe but labile
or associated with progressive deterioration of renal function or extrarenal
complications of abnormal vascular permeability, with or without
concurrent MAHA and/or thrombocytopenia.
When a patient with end stage renal disease due to AHUS undergoes
kidney transplantation, the risk of recurrent TMA and graft loss is very
high. The graft loss can be prevented with anticomplement therapy.
Therefore, the possibility of AHUS should be considered when the cause
of renal failure is unknown for a candidate of kidney transplantation.
AHUS in patients with co morbid conditions
Certain conditions such as febrile illnesses, inflammation, surgery,
trauma, or intravenous radiographic contrast agents are not known to
directly link to micro vascular stenosis or thrombosis but may trigger
complement activation and the development of TMA in patients with
defective regulation of the alternative complement pathway.
In other co morbid conditions such as hematopoietic stem cell therapy,
pregnancy, or autoimmunity with positive antinuclear antibodies,
antineutrophil cytoplasmic antibodies or antiphospholipid antibodies,
renal insufficiency with MAHA and/or thrombocytopenia may result
from AHUS or other mechanisms. AHUS is the presumptive diagnosis
after other potential causes are excluded (Table 4).
In patients who do not require immunosuppression for graft versus host
disease after hematopoietic stem cell therapy, deranged recovery of the
immune system may lead to the generation of antibodies of complement
factor H (CFH) and hence AHUS. The complications of AHUS may appear
a few weeks to several months after myeloablation or discontinuation
of immunosuppressive drugs [11,24]. Such post-immunosuppression
autoimmunity can also lead to inhibitory autoantibodies of ADAMTS13
and acquired TTP.
For most patients presenting with renal insufficiency, MAHA and
thrombocytopenia and are found to have positive antinuclear antibodies,
antineutrophil cytoplasmic antibodies, or antiphospholipid antibodies
but no severe ADAMTS13 deficiency, kidney biopsy is often needed to
distinguish TMA from other types of pathology such as vasculopathy
(e.g. renal scleroderma), vasculitis, and microvascular fibrin-platelet
thrombosis (e.g. catastrophic antiphospholipid antibody syndrome) .
During pregnancy, the HELLP syndrome is the most common cause
of MAHA and thrombocytopenia. On the other hand, activation of
the complement system during normal pregnancy [25], can trigger the
development of TMA in patients with preexisting defective regulation
of the alternative complement pathway. If such women happen to have
preeclampsia or the HELLP syndrome, the transition to AHUS may not
be easily recognized. Such cases of AHUS are often assumed instead
to be worsening of preeclampsia/HELLP. Misdiagnosis of AHUS as
preeclampsia/HELLP may account for the high prevalence (~10%)
of AHUS mutations in clinical series of preeclampsia/HELLP [26].
AHUS should be suspected when renal failure is severe or presumed
‘preeclampsia/HELLP’ persists, worsens or occurs after delivery.
Confirming the diagnosis of AHUS
The diagnosis of AHUS is confirmed by mutation analysis of regulators
of the alternative complement pathway, which includes complement
factor H (CFH), CD46 (membrane cofactor protein, MCP), complement
factor I (CFI), and thrombomodulin (THBD) and ELISA for antibodies
of CFH. Mutation analysis also includes complement factor B (CFB) or
C3, since gain of function mutations of either protein may disrupt the
regulation of the alternative complement pathway.
The laboratory tests for confirmation of AHUS are not yet optimal. The
tests only identify 40% -75% of patients that are known to have defective
regulation of the alternative complement pathway. Negative test results
do not exclude the diagnosis of AHUS. Furthermore these tests may have
turnaround times of weeks to months. Therefore, for patients presenting
with acute complications, therapeutic decisions are made in most cases
when the diagnosis of AHUS is only presumptive.
New concepts in the management of TTP and AHUS
TTP
The conventional treatment for acquired TTP is plasma exchange.
While this treatment decreases the risk of death from greater than 90% to
less than 10%, it does not address the high risk of subsequent relapses [6].
Corticosteroids, cyclophosphamide or azathioprine are not very effective
in decreasing the risk of relapse and not infrequently are associated with
potentially serious complications.
Preemptive rituximab therapy, immediately after the diagnosis of
acquired TTP is confirmed, may decrease the risk of relapse for 1-3 years
[27]. Nevertheless, relapses continue to occur thereafter. Late relapses may
be prevented by a strategy of repeated rituximab therapy guided by serial
plasma ADAMTS13 activity levels [6,28].
Hereditary TTP responds to plasma infusion at approximately 5-7.5 ml/
kg every 2-3 weeks. Most patients require maintenance therapy to prevent
unpredictable but potentially serious complications such as strokes and
progressive deterioration of renal and occasionally mental functions that
may occur in some patients who are not treated.
AHUS
Historically, AHUS has been treated as TTP with plasma exchange,
supplemented, without good basis for most cases in retrospect, with
corticosteroids, immunosuppressive drugs such as cyclophosphamide and
rituximab and even splenectomy. With such treatment, more than 50% of
the cases die or develop end stage renal disease by one year [7,29].
Anticomplement therapy with eculizumab has been shown to be highly
effective in suppressing complement activation, thereby preventing relapse
of TMA and progressive deterioration of the kidney function [30]. Many
patients gladly find their kidney function improving with anticomplement
therapy.
In patients with thrombocytopenia or extra-renal complications of
abnormal vascular permeability, anticomplement therapy is followed by
resolution of thrombocytopenia and steady alleviation of the extra-renal
complications by one week [31]. For patients with labile hypertension,
the blood pressure often stabilizes by two weeks of treatment. Thus,
when eculizumab is instituted for presumptive AHUS, lack of expected
responses practically excludes the diagnosis of AHUS and nullify the
indication of continuing anticomplement therapy, unless there is a
reason for the lack of response. Examples for lack of response include
pseudo-thrombocytopenia due to in vitro platelet clumping, concurrent
plasma exchange, and inadequate suppression of complement activation.
Adequacy of complement suppression can be assessed by total complement
test (CH50) [32]. In contrast, improvement of the kidney function may
be quite slow, spanning over the course of many months. Because kidney
injury may be irreversible, lack of renal function improvement with
anticomplement therapy does not exclude the diagnosis of AHUS.
Patients who have evidence of disease activity such as headache,
anorexia, nausea, abdominal pain or increasingly unstable blood pressures
before each biweekly dose of treatment obviously require the treatment
more frequently and the treatment needs to continue. Anticomplement
therapy should also be continued indefinitely for patients who have
frequent relapses of disease activity or progressive renal function
deterioration without the treatment. Nevertheless, historically, it is known
that some patients have stable kidney function and no relapses for years
after one episode. For such patients, indefinite long-term maintenance
treatment would unnecessarily expose the patients to the risk of fulminant
meningococcal sepsis in association of anticomplement therapy for
uncertain benefit. Nevertheless, identifying these patients a priori remains
a challenge.
One way to overcome this challenge is a strategy of trial and error.
For patients who are asymptomatic and have stable renal functions with
anti-complement therapy, the treatment may be tapered off by gradually
increasing the length of interval before each successive dose of treatment.
During periods of tapering and treatment discontinuation, the patients
should be closely monitored. Any recurrence of symptoms, hemolysis,
decrease in platelet count, or increase in blood pressure, LDH or serum
creatinine should be promptly evaluated for the possibility of relapses and
the need of reinstitution of anticomplement therapy.
Conclusions
Since the discovery of ADMTS13 deficiency in TTP and defective
regulation of the alternative complement pathway in AHUS, it has become
apparent that the syndrome of MAHA and thrombocytopenia may result
from a group of etiologically or mechanistically diverse disorders. With
a new scheme of disease classification, it is now possible to approach the
diagnosis of patients presenting with MAHA and thrombocytopenia in a
rational manner and make the diagnosis of TTP or AHUS in patients of
without MAHA and thrombocytopenia. Correct diagnosis is an essential
first step for proper management of these potentially serious diseases.