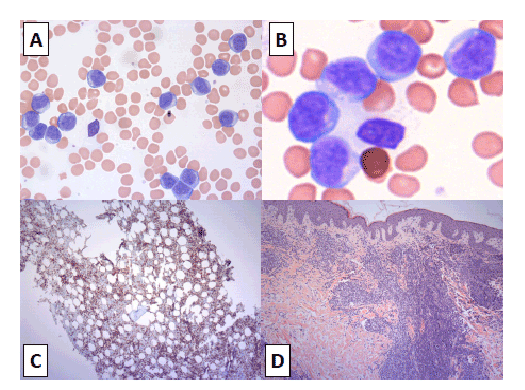
Figure 1: (A) Lymphocytes in peripheral blood, objective × 40; (B) Lymphocytes in bone marrow, objective × 60; (C) Immunostaining for CD3, objective × 20; (D) Skin biopsy, objective × 10
Rouslan Kotchetkov*1 Russell Price2
1Department of Medicine, University of Toronto, Simcoe Muskoka Regional Cancer Program, Barrie, Canada*Corresponding author: Rouslan Kotchetkov, Department of Medicine, University of Toronto, Simcoe Muskoka Regional Cancer Program, Barrie, Canada, E-mail: kotchetkovr@rvh.on.ca
61-year-old female was referred to our center for management of progressing chronic lymphocytic leukemia (B-CLL), diagnosed 4 years ago based upon lymphocytosis and smudge cells present on peripheral blood smear. At initial assessment she had erythematous papulomatous skin rash, hepatosplenomegaly, diffuse lymphadenopathy, pleural effusions. Peripheral blood (PB) showed anemia (hemoglobin 110 g/L, mean corpuscular volume 101fL), elevated white blood cell count (199 × 109 /mL) with lymphocytosis. Bone marrow examination revealed interstitial and intra-sinusoidal marrow infiltration with small to intermediate size mature lymphocytes with minimal cytoplasm, condensed chromatin and indistinct nucleoli. No cells with large nucleoli typical of prolymphocytes were seen. Cells were positive for CD2/3/4/5/7, negative for CD20/23/8, had normal female karyotype. B-cells appeared exceedingly rare. Skin biopsy and pleural fluid cytology showed similar findings. Based on morphology and immunohisochemistry a provisional diagnosis of mature T-cell leukemia/lymphoma was made. Because of the discordance between laboratory and clinical findings, additional investigations were requested, which showed that the lymphoma cells were positive for TCL-1, CD30. The diagnosis was revised to T-cell prolymphocytic leukemia (T-PLL), small cell variant, without evidence of previously diagnosed B-CLL.
Treatment/prognosis of B-CLL and T-PLL differ significantly. PB lymphocytosis should not be assumed as CLL but confirmed by flow cytometric evaluation. Diagnosis of prolymphocytic leukemia should be made based on combination of clinical, morphological and immunophenotypic features.
T-Prolymphocytic Leukemia (T-PLL); Lymphocytosis; Chronic Lymphocytic Leukemia (CLL); Smudge Cells
A 61-year-old female was referred to our cancer center for progressing chronic lymphocytic leukemia (CLL), diagnosed four years ago elsewhere. Until recently she had an indolent course, but over the last two months she had rapidly increasing lymphocytosis and generalized skin rash. At initial assessment she had ECOG status 1, normal respiratory and cardiovascular exams, generalized diffuse lymphadenopathy, limited erythematous papulomatous skin rash, and mild hepatosplenomegaly. Peripheral blood (PB) investigation revealed mild anemia (Hb 110 g/L, MCV 101fL), lymphocytosis (191 × 109 /mL) and few smudge cells. PB lymphocytes were small to intermediate size with minimal cytoplasm, condensed chromatin and indistinct nucleoli (Figure 1A). Preliminary diagnosis of CLL was made and original flow cytometry (FC) report was requested. Skin lesion was biopsied, and staging computer tomography (CT) scans were arranged. Since the patient was asymptomatic, we put her on active surveillance.
Within two weeks her condition deteriorated rapidly and she was admitted with progressive fatigue, ascites, and dyspnea secondary to large pleural effusions. Her lymphocytosis increased to 303 × 109 /mL. CT abdomen/pelvis/chest confirmed lymphadenopathy above and below diaphragm, with the largest lymph node measuring 3 cm. Transformation into aggressive lymphoproliferative malignancy (LPM) was suspected and she underwent bone marrow aspiration and biopsy. While waiting for the results of the investigations she received a short course of prednisone with no success.
Bone marrow examination revealed interstitial and intrasinusoidal marrow infiltration with small to intermediate size lymphocytes with minimal cytoplasm, condensed chromatin and indistinct nucleoli (Figure 1B). No cells with large nucleoli typical of prolymphocytes were seen. Cells were positive for markers CD2/3/4/5/7, negative for CD20/23/8 (Figure 1C; CD3 immunostain), and had normal female karyotype. B-cells appeared exceedingly rare. Skin biopsy (Figure 1D) and pleural fluid analysis showed similar findings. Based on morphology and immunohistochemistry a provisional diagnosis of mature T-cell leukemia/lymphoma was considered. She started chemotherapy with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), however her condition deteriorated after the first cycle. She remained in-patient, required chest tube with ongoing drainage and recurrent paracentesis, and skin rash progressed to approximately 75% of her body surface area.
Because of discordance between laboratory and clinical findings further investigations were requested. These showed the lymphoma cells were positive for TCL-1 and CD30. The diagnosis was revised to T-cell prolymphocytic leukemia (T-PLL), small cell variant. While reviewing patient’s original data we realized that no FC from PB was done originally. B-CLL had been incorrectly diagnosed based solely on lymphocytosis and presence of smudge cells in PB.
Treatment with Alemtuzumab, anti-CD52 antibody, was initiated and within 2 weeks she reached complete remission with complete resolution of symptoms, lymphadenopathy and skin rash. Her lymphocyte count returned to normal, hemoglobin and platelet count improved. She was referred to the allogeneic bone marrow transplantation (Allo-SCT).
Figure 1: (A) Lymphocytes in peripheral blood, objective × 40; (B) Lymphocytes in bone marrow, objective × 60; (C) Immunostaining for CD3, objective × 20; (D) Skin biopsy, objective × 10
There are several teaching points to be discussed in management of such patients.
Smudge cells (SC), also known as “basket cells” or “Gumprecht cells”, are often reported in PB smears in patients with lymphocytosis. SCs are remnants of cells that lack any identifiable cytoplasmic membrane or nuclear structure. Blasts and lymphoid cells are fragile and crush easily with mechanical pressure during the PB smear processing, resulting in SC [1]. SC is not unique to CLL, but is seen more frequently and in higher numbers in CLL than in other conditions, both neoplastic and reactive. SC of non-lymphoid origin can also be seen in degenerating samples. Normal specimens may contain 0.01% of SC, in patients with severe infections/ burns 0.1 to 0.3%, in acute leukemia’s 1 to 3%, and in CLL up to 20% or higher. Percentage of SC in PB smears is not helpful as a morphologic finding for the differentiation between CLL and other B-cell malignancies [2] but may reflect burden of the disease in patients with confirmed, early stage CLL. Presence of more than 30% SC was associated with shorter median progression-free period compared with patients who had less than 30% SC [3].
In addition to acute and chronic leukemias, including T-PLL, there are several LPM presenting with different degree of PB lymphocytosis (Table 1). Overall frequency of PB lymphocytosis in LPM, excluding chronic leukemia, is almost 30% as shown by a combination of morphology and flow cytometry [4]. T-PLL is usually associated with marked lymphocytosis >100,000/mL. A half of the patients present with absolute lymphocyte count >200,000/mL [5]. T-prolymphocytes are usually easy to recognize by morphology: small to medium-sized lymphoid cells with non-granular basophilic cytoplasm, round, oval or markedly irregular nuclei and a visible nucleolus. However, in 25% of cases cells are small with indistinct nucleolus, called “small cell T-PLL variant” [6].
In addition to blood smear review PB lymphocytosis should be evaluated by FC for presence and absence of specific antigens (immunophenotype) of individual cells. In the assessment for LPM FC provides information about cell lineages (T-/B- or NK- lymphoid cells), determination of maturity (mature T/B-lymphoid cells vs blasts), detection and documentation of abnormal cell phenotype (immunoglobulin light chain class restriction and aberrant antigen expression) and level of antigen expression (intensity of staining by fluorochrome labeled antibodies) [7]. Immunophenotype for classical and small cell variant T-PLL is identical.
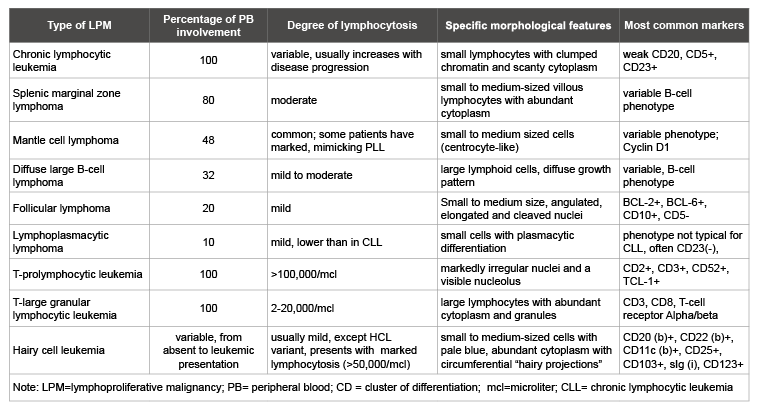
Table 1: Most common lymphoproliferative malignancies presenting with peripheral lymphocytosis
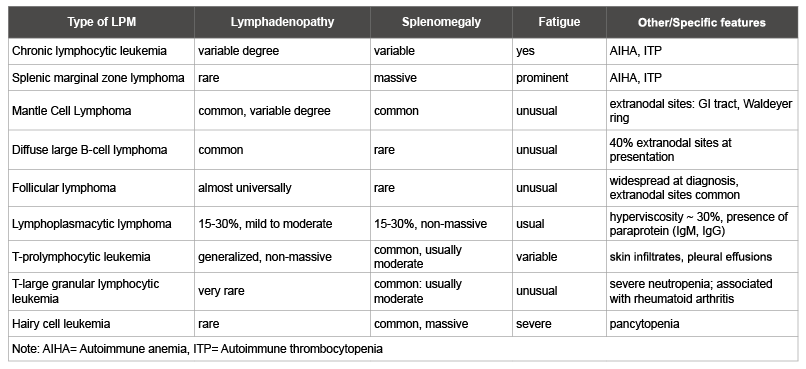
Table 2: Clinical features of lymphoproliferative malignancies presented with peripheral lymphocytosis
Clinical features of LPM presenting with PB lymphocytosis are presented in table 2. In contrast to other LPMs, T-PLL typically presents as an aggressive disease with a median survival of usually less than 1 year. Often, multi-organ involvement with non-bulky lymphadenopathy is presented early at diagnosis. Unusual in B-CLL, but common in T-PLL features include skin infiltration, nodular, macropapular rash, erythroderma, and less frequently serous effusions, mainly pleural [8]. Anemia and thrombocytopenia are common.
Approximately 15% of patients may be asymptomatic at diagnosis, and this “indolent” phase can persist for a variable length of time. Such cases may show an accelerated course after 2-3 years and in rare cases may extend to several years [9]. Therefore, diagnosis of T-PLL should be made based on combination of clinical presentation, morphological and immunophenotypic features by FC. T-PLL is usually resistant to conventional chemotherapy and can be rapidly fatal if not treated properly, thus making a correct diagnosis essential. As in our patient the clinical presentation was not consistent with B-CLL or peripheral T-cell lymphoma, therefore we pursued further laboratory diagnostic work-up. When the correct diagnosis of T-PLL was made, we switched therapy from CHOP to alemtuzumab and the patient’s condition improved quickly.
Patients with asymptomatic lymphocytosis that is stable or only slowly progressing could be on active surveillance. Since progression may occur rapidly, we suggest referral of T-PLL patients to a cancer center shortly after diagnosis. T-PLL patients should be monitored more frequently than patients with B-CLL. Thus, if needed, treatment should be initiated as early as possible. Debulking therapy with steroids has no or very limited effect in T-PLL. The best treatment is alemtuzumab, and where possible followed by consolidation with allogeneic stem transplant (Allo-SCT). We recommend an early referral to Allo-SCT team, allowing time to identify a suitable donor if Allo-SCT is planned and avoid delays once treatment is initiated.
Alemtuzumab is a humanized IgG1 antibody, which targets the CD52 antigen highly expressed on normal and malignant T- and B-lymphocytes, monocytes, and at particularly high density on T-PLL, but not on hematopoietic stem cells [10]. Single agent Alemtuzumab is efficient in T-PLL with response rate over 90% and better tolerated in T-PLL population with 4-fold fewer infectious complications as compared to relapsed CLL patient group [8]. Infection prophylaxis for Pneumocystis Jirovecci and herpes viruses, and regular monitoring for cytomegalovirus reactivation help to minimize serious infections. Sequential chemoimmunotherapy based on fludarabine, mitoxantrone and cyclophosphamide induction followed by alemtuzumab consolidation (FCM+A) also seems to offer an alternative to alemtuzumab alone [11]. Bendamustine is an original alkylating agent that shows only partial cross-resistance with other DNAbinding anticancer agents and no cross-resistance with other alkylating agents. Bendamustine is widely used in the treatment of indolent B-cell lymphomas, including CLL. Recent study of bendamustine showed its efficacy and safety in T-PLL, particularly in patients who are refractory to alemtuzumab [12].
In conclusion, PB smudge cells and lymphocytosis are not diagnostic for CLL, but should be further investigated by morphology review and FC. T-PLL is a rare, aggressive disease with unique clinical presentation, laboratory features and requires special management.
None declared.
The authors have obtained patient consent.
Rouslan Kotchetkov collected and interpreted clinical data, performed review of the literature, drafted and revised the manuscript. Russell Price acquired and interpreted pathology images for the publication, and revised the manuscript. All the authors approved the final version to be published and agreed to act as guarantors of the work.
Download Provisional PDF Here
Article Type: Case Report
Citation: Kotchetkov R, Price R (2016) Microcellular variant of T-Prolymphocytic Leukemia mimicking CLL. J Blood Disord Med 1(1): doi http://dx.doi. org/10.16966/2471-5026.104
Copyright: © 2016 Kotchetkov R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access