Introduction
When the goal of an experiment is to determine the proteins that distinguish one sample from another in complex biological samples, biochemists traditionally use 1D SDS gels or 2D isoelectric focusing / SDS gels to reveal quantitative changes in expression patterns. More recently, mass spectrometric techniques have been developed to extend beyond the tens of proteins detectable by SDS gels to thousands of proteins, often aided by using added heavy atom labeled synthetic peptides to improve quantification [1,2]. However, these mass spectrometric measurements often require extensive protein and / or peptide separations for identifications [3-8]. These separation requirements can introduce variability, reduce throughput and increase the expense of the analysis.
At the other extreme is mass spectrometry without separation, resulting in spectra with peaks corresponding to the most abundant and readily detectable peptides. In the case of a tryptic digest of a protein preparation, most MALDI peaks correspond to arginine-containing peptides. According to the scientific literature, attempting peptide mass fingerprinting (PMF) on complex mixtures of proteins is not feasible, because too few peptides would be expected to derive from any particular protein. Recently, however, advances in MALDI mass spectrometers have enabled accurate mass measurements to within a few parts per million (ppm). Increased mass accuracy decreases significantly the number of peptide sequences that can account every peak, consequently increasing the usefulness of PMF [9].
This manuscript aims to show that it is possible to identify correctly the major proteins in complex mixtures by PMF. Using tryptic digests of chicken muscle as a model system, it is also possible to distinguish correctly between closely related protein isoforms like myosin heavy chains, and to determine some quantitative changes in abundant proteins. To accomplish this, we employed a PMF program optimized for MALDI MS data. This program takes explicit advantage of the higher detectability of arginine-containing peptides [10], the high mass accuracy that can be obtained with today’s mass spectrometers, and the peak intensities of the mass list. Typical mass spectra of complex mixtures contain several hundred peaks between m/z 800 and 4000 following reduction of isotope clusters to mono-isotopic masses. In the first round of PMF, the most abundant protein is identified, which generates a set of masses that can be used for internal calibration. At this stage, biological information about the sample should be used to ensure that an appropriate protein is chosen for calibration. In chicken muscle samples, in accordance with biology, the most abundant protein is actin. Following internal calibration on actin peptides, the mass tolerances can be tightened, enabling the identification of additional proteins. Using these methods, we demonstrate that it is possible to identify small proteins based on as few as two peptides, provided the peptides contain arginine and are expected terminal digestion products of trypsin.
The chicken muscle system was chosen because chicken muscle samples are readily available from grocery stores. If PMF is successful on chicken meat, then it should also be applicable to human muscle biopsy samples. It is well established that the patterns of myosin heavy chain isoforms become altered upon muscle pathology, often due to changes in underlying muscle fiber usage [11]. However, when dealing with human tissue, strict clinical guidelines must be followed prior to experimentation. In the chicken system, it is well known that there are two kinds of meat, white meat and dark meat. In particular, white meat (e.g. pectoralis major muscle) has more fast muscle fibers, and dark meat (e.g. adductor superficialis and lateral gastrocnemius) has more slow muscle fibers [12-15] and more myoglobin, which is thought to be directly responsible for the color of the meat [16]. Moreover, in proteomic analyses, it is often considered invaluable to obtain freshly excised tissue, because protein breakdown may confuse the protein identification process [17]. If protein identification by PMF proves feasible starting from grocery chicken meat, then the identification process ought to be robust enough so that any degradation that takes place in human muscle biopsy samples will not be a limiting factor for isoform identification. Finally, the chicken proteome is fairly well characterized [18], which is a necessary requirement for PMF to succeed.
Experimental Procedures
Preparation of Tryptic Digests from Muscle Extracts
Four samples of dark meat from chicken thighs (adductor superficialis and lateral gastrocnemius) and three samples of white meat (pectoralis major) were obtained from a local supermarket.
Each sample was from middle sections of the muscle, not near cartilage, tendon or skin. Samples (approximately 100 mg) were homogenized in 1 mL of 100 mM Tris pH 8.0 containing 1% SDS and 10 mM dithiothreitol (DTT) , heated to 90°C for several minutes, cooled to room temperature, and alkylated with 25 mM iodoacetamide. Excess alkylating agent was quenched with 100 mM mercaptoethanol. About 10% of the sample was then precipitated in acetone, which removes to the supernatant most salts, nucleic acids and lipids. The dried pellet was digested with bovine trypsin (Sigma) at about 200:1 substrate protein: trypsin at 37°C overnight.
The trypsin to protein substrate ratio is not crucial so long as most substrate protein is digested, and so long as trypsin auto-digestion peptides remain invisible, or nearly invisible in the MALDI spectra. Under these conditions, peptides containing methionine are recovered predominantly in the reduced methionine form, and actin peptides derived from chymotryptic-like contamination by cleavage following tyrosine are not detectable. If these two objectives are achieved by appropriate digestion protocol adjustments, the spectra interpretation is simplified.
Gathering of MALDI spectra
Digests were diluted into 5 mg/ml alpha-cyano hydroxycinnamic acid (HCCA) dissolved in 75% acetonitrile containing 0.1% trifluoroacetic acid, and spotted in duplicate on MALDI plates. Spectra were internally calibrated on the major tryptic peptides from actin, which are among the most intense in any tryptic digest of unseparated skeletal muscle proteins. The highest quality spectra are typically acquired at an intermediate dilution of the peptide digest, where the goal is to acquire spectra with the largest possible set of well resolved isotope clusters.
Spectra were acquired on a custom-built MALDI reflector instrument with a 7.5 m flight tube manufactured at Virgin Instruments, and collected in duplicate. Data were collected every 1 ns at a laser frequency of 1 kHz using a laser pulse current of 2.4 and a focus mass of 1500 Daltons; with a scan speed of 1 mm/s over a mass range of 10-2200 Daltons (35 -530 microseconds). The analyzer region was pumped down to a pressure of 2 × 10-8 Torr. Typical averaged spectra from 5000-6000 laser shots are shown in figure1. This averaging process takes little additional MS acquisition time but improves the mass accuracy and isotope envelope intensities of the minor peaks.
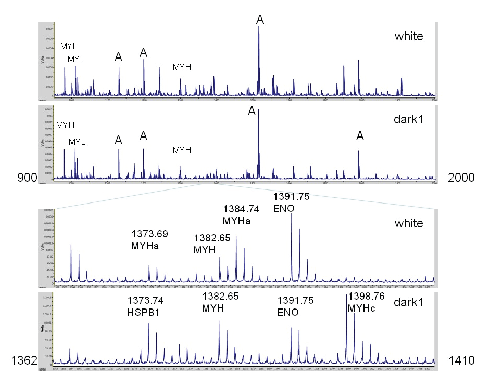
Figure 1: Comparison of Spectra from White Meat to Dark Meat. The top two traces show masses between m/z 900 and 2000. Seven prominent masses are labeled that are intense in all spectra: A, actin; MYL, myosin light chain; MYH, myosin heavy chain (shared across most isoforms). The lower traces highlight a region with prominent differences due to myosin heavy chain polymorphisms. In the 3rd trace, the peak at 1384.74 corresponds to a myosin heavy chain peptide diagnostic of white meat. The corresponding peptide from dark meat appears at 1398.76. The peaks at 1391.85 and 1382.65 correspond to enolase (which is more abundant in white meat) and a myosin heavy chain peptide that is widely shared. In the top spectrum, the 1373.69 peak corresponds mostly to myosin heavy chain. A peak with a similar mass (at 1373.74) from heat shock protein B1 is more prominent in dark meat.
PMF
Virgin Instruments SimulTOF software was used at the first stage for all PMF experiments. This software has built in averaging and peak list de-isotoping functions. It also has a built-in PMF program based on the ChemPlex software [19] (VChemplex). First, each theoretical peptide is assigned a chemistry score based on its sequence and flanking residues. Arginine-containing peptides with no missed cleavages and no compromising flanking residues (e.g. acidic residues) are assigned a score of 20, whereas the highest score for a lysine-containing peptide with no arginine is 2. VChemplex first screens for proteins with at least one arginine-containing tryptic peptide with no missed cleavage that maps to within 4 ppm of any of the 100 most intense peaks. This level of mass accuracy requires internal calibration. When a protein passes this first constraint, the program next requires matching of at least one additional peptide to within 10 ppm containing up to one missed cleavage from the complete peak list. The protein score that is calculated is proportional to the percentage of peak intensity that can be accounted for by the protein, proportional to the percentage of expected tryptic peptides that are mapped, where each peptide has a numerical score for matching based on its sequence and flanking residues, and inversely proportional to the intensity-weighted average ppm for the matches [19]. The initial list of proteins and matched peptides, often containing thousands of proteins, is then filtered to include only proteins that have matched peptides whose total chemistry score accounts for least 20% of the total possible chemistry score for all of the peptides in the protein. This drastically reduces the proteins to be considered further. The remaining list of proteins is sorted by decreasing overall score, and the protein list undergoes a process termed iterative subtraction. The score of each protein is recalculated but the masses in the peak list become unavailable for matching by lower ranking proteins, if there is a match to a credible peptide from a higher ranking protein. The iterative subtraction process ensures that when multiple isoforms of a protein (like myosin) are present that share common peptides, there will always be one ‘winning’ isoform. Each peak list, which typically contained about 400 deisotoped peaks, was searched against a combined Swiss-Prot and TrEMBL database containing 15148 protein sequences. This combined database was downloaded from the UniProt web site (http://www.uniprot.org/), and reconfigured into a sqlite3 database for use with VChemplex. The input for VChemplex is the MALDI spectrum displayed to the user (using the SimulTOF viewer), and is dependent on the peak detection settings selected by the user. The deisotoping function within the SimulTOF viewer maps the area of each isotope cluster to the mono isotopic mass in the peak list. Online Resource Table S1 contains a list of all of the settings used here for protein identification.
To address the question of false positives, each spectrum was also subjected to PMF using a database that contained human and zebrafish sequences in addition to chicken (204,304 protein sequences). When this larger database was searched, of the 1400 queries (100 peaks from 14 samples), only 678 peptides were assigned correctly (see column “same MSMS”) compared to 776 when the smaller database was searched. To our surprise, 27 chicken peptides were identified by PMF that were consistent with MS/MS data that had not been identified when the smaller chicken database was searched (highlighted in blue in column “SeqLarge”), presumably because a human or zebrafish protein ‘took out’ some masses that otherwise would have been matched to an incorrect chicken protein. Based on these results, it would appear the majority of these identifications are correct, and that the MS/MS data provide a useful means to ascertain correct identification.
PMF Database
Following identification of abundant chicken peptides by MS/MS (see below), some sequences were altered in the sqlite3 protein database to correspond to well-known stoichiometric chemical modifications. This facilitates subsequent peptide and protein identifications by PMF, because the VChemplex program will then score a particular protein isoform higher if the expected modification is present, and lower the score if the expected chemical modification is absent. Accordingly, expected changes to the N-terminus of prominent chicken muscle proteins, e.g. changes at the N-terminus of mature proteins due to acetylation (actin and myosin) and removal of signal sequences, were entered into the protein sequence. One histidine residue in both actin and myosin heavy chains was similarly converted to methyl-histidine in all prominent isoforms, based on annotations in the Swiss-Prot database and MS/MS identifications.
Separation of Peptides by HPLC
Aliquots of the digested muscle samples were injected onto an HPLC system using a 150 × 0.3 mm Prot 200 C18 5 µm column (The Nest Group, Southboro, MA), and separated by gradient elution. Buffer A was 2% acetonitrile/ 0.1% TFA, and Buffer B was 85% acetonitrile / 5% isopropanol / 10% water/ 0.1%TFA. Gradients had 3 segments, from 2-10% Buffer B over 5 min, 10% to 45% Buffer B over 60 (or 130) min, and 45% - 100% Buffer B over 10 min with a flow rate of 4 µl/min. Matrix (4 µl/min, 5 mg/ml HCCA in 75% acetonitrile/0.1% TFA and 10 picomole/µl synthetic peptide standard) was added before spotting every 5 sec onto 384 spot plates. Note these separations were performed for supporting MS/MS analysis only.
Acquisition of MALDI spectra
MS spectra were collected on prototype mass spectrometer with a 7.5 m flight tube from m/z of 10 to 2200 every ns (∼35 microseconds to ~530 microseconds) at 1 kHz. The upper m/z limit of 2200 was necessary due to the capacity of the digitizer, because collection started at 10 m/z. Spectra could have been acquired by starting at m/z 500 (or even 800), and would have resulted in a larger number of peaks matched by PMF. Typically, 5000 spectra were averaged together, resulting in a resolution of 35000. Spectra were internally calibrated on actin. MS/MS spectra were collected on a SimulTOF-300 with a source voltage of 1 kV and a second source voltage of 2 kV. Typically, 5,000-20,000 shots were collected per precursor, with up to 10 precursors per spot.
Informatics
Fingerprinting was performed using the VChemplex program, which was built into SimulTOF software. Search parameters metadata and peak lists, peptide and protein identifications are downloaded into a SQLite3 database. To speed up SQL queries, theoretical masses and peak m/z were first mapped to their nearest mass bin, calculated by rounding the mass divided by 1.0005. This enables first pass integer-based alignments.
PCA analysis:
To perform a PCA analysis, the data must be organized into a matrix of mass features vs. sample that is appropriately normalized. The following steps were performed to obtain this matrix. For the experiment described here, two spectra were acquired from seven different digests of muscle. Following PMF, the output was deposited into the SQLite3 database, and then imported into Microsoft Access. Masses that correspond to standards or non-peptides were removed based on their mass defect. Each peak was then assigned an intensity rank for each spectrum. The resulting table of 1400 masses (Table S2) was next grouped into mass bins, and the standard deviation of each mass bin was determined. In this dataset, there were seven mass bins that were manually split into two mass features because the standard deviation of the masses was greater than 0.025. At this stage, the combined mass list was filtered to contain those mass features that were encountered at least 3 times across the 14 samples. In these data, this resulted in 157 mass features, ranging in mass between m/z 800 and 2115. To prepare the mass features for PCA, the 157 peaks and their raw intensities were copied into excel, and then doubly normalized, first by column, and then by row (converting each intensity into a percentage). A comma delimited file consisting of these data was imported into R (see Table S3, columns “Peak” and columns “1a-7b”), and PCA analysis was performed using the prcomp function in the R statistical package (http:// www.r-project.org/). The principal component table from R was then exported back into excel for plotting purposes.
Results
Spectra of digests of meat extracts
As seen from these spectra, most of the strong peaks are shared. The insets show the region between m/z 1362 to 1410. Some of the peaks are strong in one spectrum, but weak or nearly absent in the other; for example the peaks at 1384.74 (strong in white meat) and the peak at 1398.76 (strong in dark meat, but weak in white meat). The differential peak intensity demonstrated by the insets in figure 1 is not restricted to this particular mass region. To establish the reproducibility of these patterns, additional samples from white and dark meat was prepared at different times originating from different trips to the grocery store. In all cases, differences between white and dark meat were readily apparent, suggesting that some abundances of some proteins were dramatically different between white and dark meat. Follow-up experiments were performed that consisted of three samples of white meat and four samples of dark meat, each of which was spotted in duplicate on the MALDI plate, resulting in 14 spectra that are described in detail below.
Ida |
Sampleb |
Categoryc |
Peaksd |
Top 100 Peakse |
|
|
|
PMFf |
Correctg |
MSMSh |
1 |
1a |
w |
420 |
58 |
53 |
72 |
2 |
1b |
w |
420 |
61 |
58 |
74 |
3 |
2a |
w |
414 |
54 |
50 |
67 |
4 |
2b |
w |
420 |
53 |
47 |
70 |
5 |
3a |
w |
420 |
56 |
52 |
74 |
6 |
3b |
w |
420 |
53 |
50 |
68 |
7 |
4a |
d2 |
420 |
66 |
55 |
76 |
8 |
4b |
d2 |
420 |
58 |
50 |
75 |
9 |
5a |
d1 |
415 |
59 |
52 |
71 |
10 |
5b |
d1 |
420 |
61 |
55 |
73 |
11 |
6a |
d1 |
381 |
58 |
52 |
74 |
12 |
6b |
d1 |
395 |
63 |
52 |
71 |
13 |
7a |
d2 |
385 |
66 |
62 |
76 |
14 |
7b |
d2 |
357 |
63 |
60 |
78 |
average |
|
407.6 |
59.2 |
53.4 |
72.8 |
sum |
|
5707 |
829 |
748 |
1019 |
Table 1:Samples
Samples aID, the index number for the sample. bSample, a key describing the biological digest. cCategory, the category to which the sample belongs, as deduced by PCA analysis, as shown in Fig. 2A. dPeaks, the number of peaks used in PMF fingerprinting. The maximum number of 420 is achieved if there are 30 peaks in each 100 amu increment. eTop 100 Peaks: fPMF, the number of peaks in the top 100 that are matched to any of the top 10 proteins listed in Table S4 whether supported by MS/MS data or not. gCorrect, the number of those peaks that are consistent with MS/MS analyses. hMSMS, the number of peaks in the top 100 whose identity can be inferred from MS/MS analysis performed following LC /MALDI analysis of similar chicken muscle tryptic digests. More peptides can be identified by MS/ MS than can be assigned correctly by PMF alone, and these peptides are highlighted in column M of Table S2.
PMF analysis
The Table 2 shows that 53 peaks were matched to myosin, and 12 peaks were matched to actin (column PepI). Fifty-one and 10 of the peaks (column Pep) that mapped to myosin and actin passed criteria that judged them to be suitable for iterative subtraction because they mapped to terminal digestion products of tryspin, or missed cleavage peptides with flanking residues that trypsin is known to be less efficient at cleaving [20]. Accordingly, the corresponding masses were removed from the peak list prior to calculation of the score for the proteins that ranked lower in the table. Table 2 also lists the score (ScoreI) that the protein would have received starting from the initial list, had all peaks been available. In the case of the protiens ranking 3rd and 7th in the table, the score improves slightly with fewer matched peaks because the remaining peaks match more accurately, and therefore the intensity-weighted ppm match average, which contributes to the score, is slightly lower. In table 2, the protein names are colored according to protein category, which emphasizes that the top protein hits correspond to actin and myosin subunits. Table S4 is an extended version of the same PMF analysis as shown in table 2. It contains the top 25 proteins that were identified from each mass spectrum (14 × 25 proteins in all, many redundant). Upon examination of the names of the lower ranking proteins in this table, it is apparent that after 10 rounds of iterative subtraction, most lower ranking proteins are incorrect, as they do not correspond to known abundant proteins (which are colored according to category), and apparently random proteins appear starting from closely related samples. Nonetheless, occasional proteins in the 11- 25 range are probably correct, based on supporting MS/MS data. Table S2 lists the sequence that was assigned to each of the top 100 peaks for each of the 14 spectra (1400 peaks matched). After 100 rounds of iterative subtraction, most of the peaks get mapped to a protein, but a few remain unassigned because the mass cannot be mapped to any tryptic peptide deriving from a protein with at least one terminal arginine-containing peptide that matches to one of the top 100 masses within 4 ppm. The proteins corresponding to each sequence are colored using the same scheme as in table S4.
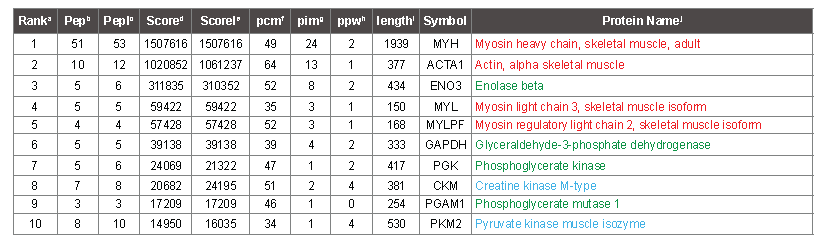
Table 2: Top 10 protein hits to sample 1 of white meat, deriving from one mass spectrum. aRank, the priority order of protein identifications, sorted according to column Score. bPep, the number of peptides that map to the peak list, after iterative subtraction. cPepI, the initial number of peptides that map to the peak list, prior to iterative subtraction. dScore, the score for the protein, after iterative subtraction. eScoreI, The score in the absence of iterative subtraction. fpcm, the percent chemistry score matched of the peptides (following iterative subtraction) that map to the protein. gpim, the percent intensity matched for the protein. hppw, the intensity weighted average ppm deviation between the measured peak masses and the calculated peptide masses. ilength, the length of the protein in aa. jSymbol, a protein abbreviation related to the nomenclature used to designate the gene encoding the protein. kProtein Name, a convenient name for the protein, adjusted for clarity. Protein names in red indicate actin or myosin subunits; green indicates glycolysis, blue indicates other abundant muscle proteins confirmed by MS/MS.
MS/MS confirmation of PMF results
To verify the results of the PMF protein identifications, and to validate the usefulness and accuracy of our PMF workflow, MS/MS data were gathered on more than 700 distinct peptides following LC separation. By studying the intact protein sequences, the top 10 proteins from the 14 fingerprints could be consolidated down to 26 protein isoforms (Table 3). Of these 26, 21 were verified by MS/MS spectra, based on at least 3 distinct MS/MS identifications. All of these 21 corresponded to abundant muscle proteins (column MSMS). Four of the other 5 were close to the bottom of the list (>=7), and are singleton identifications that are most likely spurious as they are not recognizable as abundant muscle proteins. The remaining singleton identification was a myosin heavy chain isoform (MYHg, atrial) that upon inspection did not contain any peptides supported by MS/MS that were not also attributable to at least one of the other myosin heavy chains. Therefore, six myosin heavy chains are supported by MS/MS data based on 4 or more peptides that cannot be attributed to a smaller set of myosin heavy chain sequences. At least one MYH N-terminal peptide is assigned by PMF in each of the 14 spectra, which accounts for 4 different MYH isoforms. From this observation, it is clear that the most abundant myosin heavy chain differs between the seven chicken meat samples that have been analyzed. At the peptide level, many but not all of the peptides assigned by PMF are corroborated by MS/MS, and are highlighted in green in column “Sequence” in table S2. On the other hand, as expected, many peptides that were identified by MS/MS were not correctly assigned in the PMF spectra (highlighted in violet in column “SequenceMSMS” in table S2, and marked “incorrect” or “not in database” in column “conclusion”). See the table S2 legend for more details regarding the relationship of PMF assignments and MS/MS identifications. Some of the peptides identified by MS/MS are not evident at all among any of the mass signals in any of the PMF spectra, presumably because they are suppressed at the level of ionization.
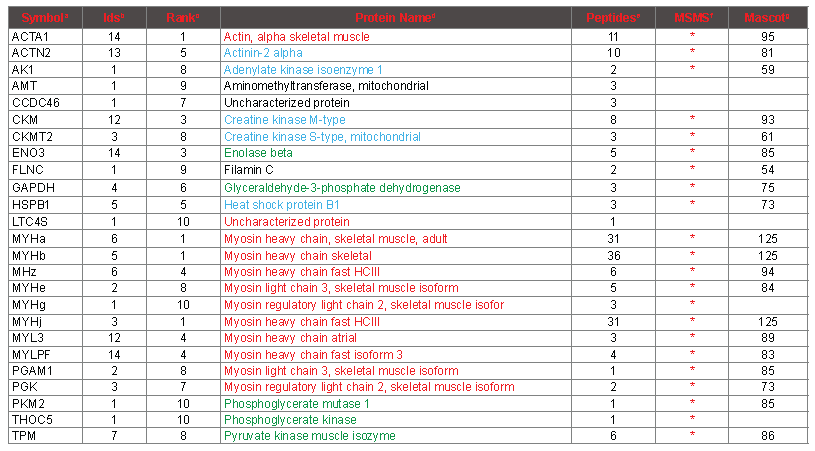
Table 3. Consolidated Proteins Identified by PMF.
aSymbol, an abbreviation for the protein. bIDs, the number of samples from which the protein
was identified in the top 10 by PMF (maximum 14). cRank, the lowest rank at which the protein was identified among the 14 samples. dProtein Name, a common descriptive name for the protein. Coloring scheme: Actin and myosin in red, glycolytic enzymes in green, other abundant muscle proteins in blue, incorrect identifications not colored. ePeptides, the maximum number of peptides of the top 100 attributed to the protein by PMF from any one sample. fMSMS, “*” indicates that peptides specifi to the protein were identifi by MS/MS. gMascot, the highest mascot score for any one peptide obtained by MS/MS.
PCA analysis
Using principal component analysis (PCA), one can determine how separable the samples are from one another without regard to identification of the masses. To accomplish this, a table was prepared containing all of the mass features that ranked within the top 100 masses in at least 3 of the spectra with an internal mass consistency of 15 ppm, which results in a table containing 157 mass features (Table S3), including 7 pairs of masses within 1 amu (see Table S5). Forty of these mass features were found in all 14 spectra at some intensity (column N), whereas at the opposite extreme, 23 features were present in each of 3 spectra. The intensity data were doubly normalized and subjected to PCA analysis, as described in Methods (Columns “1a-7b”). Figure 2A shows that PCA cleanly separates the 14 samples into 3 categories: one category of white meat, and two distinct categories of dark meat.
PCA analysis also reveals which mass features are responsible for the clustering, and this is independent of the identification data. As described above, many of the masses in table S2 have been mapped to specific proteins, which make it possible to label selectively the mass features based on the proposed identifications. In figure 2B, the masses that were mapped by PMF and MS/MS to invariant proteins like actin (11 peptides, see table S3 column ‘Symbol’), myosin light chains (4 peptides), and peptides shared among most MYH isoforms that are expressed (14 peptides) were combined into category ‘ACT’ (for actin), and are labeled with large black spots. These peptides concentrate in the center of the PCA plot, as expected for unchanging peptides. Peptides that map to any of 7 different glycolytic enzymes (15 peptides) are colored in yellow. Peptides specific to myosin heavy chain isoforms MYHa (8 peptides), MYHb (12 peptides), and MYHc (5 peptides) are colored in blue, red and green, respectively. The remaining 88 peptides are marked as small black dots. Some of them have been identified, and they are all listed in table S3. Table S6 indicates how the most readily detected myosin heavy chain tryptic peptides (as confirmed by MS/MS analysis) are shared among the 10 myosin isoforms in the TrEMBL database to which they can be mapped (listed in Table S7). No homologous set of tryptic peptides have unique peptide sequences for all 10 of these isoforms; however, there are many peptides that are distinct between the first 3 isoforms. The table shows by color-coding how prominent the mass signal was observed in the peak lists (obtained after automated de-isotoping) derived from the seven samples and 14 PMF spectra. Cells are colored according to the intensity rank of the matched peptide; if the peptide was in the top 25, it is colored red, between 25 and 50, colored orange, between 50 and 80, colored yellow. It is clear that the 3 white meat samples nearly always have higher expression of the tryptic peptide that maps to the MYHa isoform, whereas the dark1 meat sample maps best to MYHb peptides, and dark2 meat samples map best to MYHc and d peptides. This pattern is consistent with the PCA analysis in figure 2B. There is no evidence for detection of peptides unique to the MYHe - MYHj isoforms either by PMF or by MS/MS.
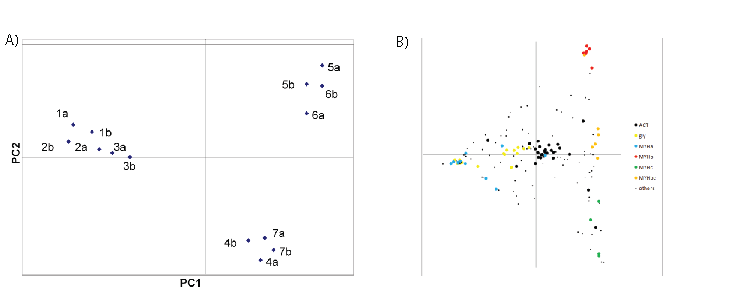
Figure 2: PCA Analysis. 157 masses with m/z between 800 and 2200 were binned by mass to within ~30 ppm, and the % intensity distribution across the 14 samples was calculated. Blank mass/sample elements were assigned a value of 0.01. Each element was normalized by percentage first by sample, and then by mass. The 157 × 14 matrix was subjected to PCA analysis using R, both so as to cluster by sample, and by transforming the matrix (mass vs. sample → sample vs. mass) to cluster by mass. The sample clustering is shown in Fig. 2A. The samples sorted into 3 different groups; white meat separated from dark meat along PCA component 1, whereas the dark meat was separated into 2 groups along PCA component 2. Upon mass-based PCA analysis, the masses were filtered and colored according to protein categories to which they had been mapped in Fig. 2B. 29 peptides shared among nearly all myosin isoforms, from actin, and from myosin light chain MYLPF were pooled together into the category labeled “ACT”, and colored black. These peptides congregate around the center of the PCA plot. Myosin heavy chain isoformspecific peptides were split into four categories; MYHa (blue, 11 peptides), MYHb (red, 7 peptides), MYHc (green, 4 peptides), MYHbc (orange, 6 peptides) and correspond roughly to the symbol column in Table S7. 15 peptides from 7 different glycolytic enzymes are marked “gly” and colored yellow. The 85 remaining masses were assigned to the category “others” whether or not they were identified by MS/MS or assigned by PMF. The category assignment of each peptide is listed in column PCA of Table S6.
Discussion
PMF was performed on tryptic digests of proteins derived from the distinctive white and dark meat muscle of chickens. The emphasis of this study is to establish a comparative proteomic paradigm based on protein identification by PMF, and to determine the ability of this PMF workflow to deduce both protein identification and protein abundance. For that reason, the supplementary tables have been prepared that delineate in detail which peptide identifications appear to be correct by comparison to MS/MS data on similar samples.
When peak lists consisting of 20-40 of the strongest peaks from tryptic digests of chicken muscle are submitted to any PMF program, an actin isoform is likely to be the top hit, because actin is relatively small (377 aa long) and very abundant. When multiple protein assignments are enabled, it is sometimes possible to identify myosin as well, but because myosin is a much larger protein (~1940 aa long), larger peak lists are required to establish enough coverage for identification. Traditionally, proteomics researchers have considered that successful identification of actin and myosin from a whole tissue digest is all PMF is capable of, and that a larger peak list would increase false positive identifications at the expense of correct identifications. Yet, in these samples, at least one peak is detectable above background at every mass, which means that it is possible to get a much larger peak list. It is a challenge to reduce potentially overlapping isotope to the best monoisotopic mass and intensity list. The PMF program VChemplex was designed to take advantage of the information in large peak lists, but this requires careful adjustment of search parameters. The optimal search parameters result in the best discrimination between proteins known to be present in the sample, like actin and myosin, and proteins known to be much less abundant or absent from the sample, e.g., any protein not identified by MS/MS. Upon optimization, subtle changes to the search parameter choices affect the results; however, the top protein identifications are stable to a wide variety of parameter values (data not shown). Therefore, changes to most parameters only affect the identification of less abundant proteins. Because peak intensity and peptide chemistry play a crucial role in protein scoring, the absolute number of peaks in the peak list has a minor impact on protein scoring. The settings listed in table S1were chosen because they produced the most credible and consistent results for all 14 samples described in this manuscript, and for other chicken muscle digest samples (data not shown).
When a PMF search is performed, a calibration file can be generated based on the top protein hits that can then be used for internal calibration of the mass spectrum. In our experiments, the best results were obtained when the calibration model was based on multiple peptides from actin and myosin. This calibration model was then applied to all spectra in a given analysis by using the actin peptide at m/z 1790.8926 as a one point internal calibrant. Typical peak lists from unseparated protein digests contain several hundred to a thousand peaks between m/z 600 and 6000, but in this paper the mass range studied was between 800 and 2200, resulting in about 400 peaks (see Table 1). When a mass can be mapped to within 4 ppm with a sequence that must be an arginine-containing peptide that is also a terminal digestion product of trypsin, there are often fewer than ten tryptic peptides that can account for the peak in the chicken database consisting of 15148 protein sequences.
In order for PMF identification to be useful for characterizing muscle fiber type, it is important to distinguish which myosin isoforms are present in greatest abundance [11]. It is well documented that many homologous isoforms are highly expressed based on identifications of muscle samples from different animals using a variety of proteomic methodologies [12,14,15,20]. Moreover, according to gene chip experiments, a large number of myosin isoforms are commonly expressed in the same muscle at the same time [12,21]. This is a challenge for PMF, because many tryptic peptides are shared between several homologous myosin isoforms (see Table S6). The VChemplex program is designed to identify many proteins simultaneously from unseparated protein digests, which requires not only high mass accuracy but also careful attention to the chemistry of the peptides that are being matched. Because the VChemplex PMF program performs rounds of iterative subtraction, one myosin isoform will always out-compete the other isoforms. In our experiments, the VChemplex program identified in either 1st or 2nd place one particular myosin heavy chain isoform from white meat. This result is based largely on three terminal trypsin digestion peptides that contain arginine, namely the Nterminal peptide, the peptide starting at aa 1424, and the peptide starting at aa 1681. The latter two peptides are prominent enough in the PMF spectra to account for the PCA separation of dark meat from white meat in figure 2. Two of these three peptides (N-terminal, and aa 1681) are also polymorphic between the dark meat myosin isoforms that predominate in the muscle samples analyzed in this manuscript.
There are several ways to assess the credibility of the PMF identifications. In one method, when the peptide masses for tryptic proteins are calculated, each protein is entered in duplicate. In the second ‘shifted’ form, each tryptic peptide mass is shifted by an offset parameter. Any match to the ‘shifted’ protein must be spurious. One can also search inappropriate databases (like bacteria) together with a relevant species to test the robustness of a result. Alternatively, one can split each spectrum in two, and search using the lower masses only (e.g. between m/z 800 and 1200), and compare the results to a search using masses between m/z 1200 and 4000). In a robust identification, the same proteins are obtained from each half of the peak list. Obviously, proposed identifications can be corroborated by MS/MS experiments, either directly or indirectly following LC peptide separation (as in this manuscript). Finally, because only the most abundant proteins in a preparation can be detected by PMF, the proposed identifications should make biological sense
Although myosin derived peptides are sufficient to explain the separation of muscle samples into three groups by PCA, there are clearly many other peptides that also contribute, some of which remain unidentified. From Fig 2B, it is evident that most peptides derived from glycolytic enzymes are prominent in the white meat samples, which is characteristic of fast muscle fibers [22]. Surprisingly, myoglobin (MB), which is usually thought to be responsible for the color difference between dark and white meat [16] was not detected by PMF. MB has three arginine-containing tryptic peptides (which were all detected by MS/MS), none of which are apparent in any of the PMF spectra even upon close inspection. Thus myoglobin appears not to be abundant enough to be detectable by fingerprinting from unseparated whole muscle digests. Although the number of samples that have been examined herein is small, there is evidence of differential expression of several other proteins; for example, by both PMF and by mapping to MS/MS data, Filamin C is detectable only in white meat, while creatine kinase S-type, creatine kinase (CKMT2) and heat shock protein B1 appear to be enriched in the “dark2” samples of dark meat, which may well correlate with specific myosin heavy chain usage (isoforms c and d). It would be interesting to perform PMF analyses using the technology described here starting from single muscle fibers that are subjected to electrophysiological analyses to confirm this kind of observation.
Another limitation of PMF in performing this kind of analysis is the resolution of the mass spectrometer. The prototype mass spectrometer used here was able to produce mass spectra with a typical mass resolution of about 35000. Experiments using the lower resolution SimulTOF 2000 mass spectrometer, which has a resolution of about 15000 on complex samples like these, indicate PMF can still identify the same myosin heavy chains as was determined in these experiments, but it becomes significantly harder to identify less abundant proteins, because of unresolved parent masses. Obviously, a much larger number of proteins can be quantified much more accurately by classical proteomic methodologies using extensive peptide separation and isotope enrichment strategies, multiple reaction monitoring strategies, or 2d gel electrophoresis. However, these strategies require careful attention to reproducibility of sample preparation, and are both time-consuming and expensive. The strategy used here would be ideal for working out protocols for more careful proteomic analyses, and for screening purposes to find samples that may warrant additional attention.
In examining the proposed identifications by PMF as represented in table S4, it is clear that PMF is often able to correctly identify 10 proteins from muscle. At the peptide level, PMF correctly accounts for 787 of the out of 1400 masses in Table S2. In 381 cases, no peptide has yet been identified by MS/MS that could explain the peak, and therefore these masses are marked ‘unidentified’ in column MSMS in Table S2. It is clear from table S4 that some strong mass signals are never correctly assigned by PMF, and some of them are never identified by MS/MS either. There are at least two major reasons for this. One problem is unexpected chemical modifications. In these experiments, we altered the sequence of actin and myosin (and some other proteins) so that the VChemplex program could identify known modifications at the N- termini, and the known site of methylation of histidine. Upon MS/MS fragmentation, these latter peptides yield a strong methyl-histidine immonium ion peak at m/z 124 (data not shown). It appears that each of these modifications is nearly constitutive, in that no unmodified peptides were found. Corresponding modified peptides were previously encountered on a much larger proteomic analysis of human muscle, in which data was acquired by MS/MS using electrospray [20]. Therefore, these modifications appear to be conserved between chickens and humans. Another interesting modification is trimethylation of a lysine residue in myosin, resulting in ATDTSFK(42.046)NK at m/z 1053.557, which is conserved at the tryptic peptide level in 12 different human myosin heavy chains in Swiss-Prot. As it happens, there is a second plausible conserved myosin peptide that can account for nearly this same mass (RHLEEEIK with m/z 1053.5694), which was not identified by MS/MS among the limited amount of MS/ MS spectra that we acquired. Thus, in this case, PMF mapped this peak to the right protein for the wrong reason. As it turns out, there are only 9 cases of ambiguous mass bin assignments that could match to two distinct terminal arginine containing peptides that were confirmed by MS/MS (Table S5). As a final informatic complication, in some muscle samples (perhaps adjacent to tendon), collagen may well be sufficiently abundant so that some hydroxyproline-containing peptides could explain unidentified masses.
From the MS/MS data, it is evident that the TrEMBL chicken database is missing some key proteins, including intact versions of some isoforms of fructose bisphosphate aldolase, glycogen phosphorylase and carbonic anhydrase that are very abundant in muscle. Many of these proteins are highly conserved across vertebrate species, such that the peptides identified by MS/MS are shared with humans and other mammals, and were accordingly identified by Mascot when vertebrate databases were searched. In addition to these well-studied enzymes, muscle tissue expresses some very large proteins like titin (33423 aa long) and nebulin (6669 aa long), which could account for many signals. Titin, nebulin and other abundant proteins like tropomyosin are also extensively differentially spliced. The TrEMBL chicken proteome does not include complete sequences for each of these splice variants. Based on early genetic data, it was originally believed there were as many as 31 chicken myosin heavy chain genes [23], but there may be no more than ten skeletal muscle myosin genes [13]. At least 5 distinct myosin heavy chain genes have been isolated from adult chicken fast muscle [12]. At this point, it is not clear whether all relevant myosin isoforms are faithfully represented in the chicken proteome. Some improvements to the chicken proteome may have been made while this manuscript has been prepared.
In some cases, masses that were prominent in PMF spectra do not seem to be prominent following peptide separation. Additional work will be necessary to determine whether this lack of correspondence can mask additional informatic limitations. There were 39 masses found in at least 4 of the mass spectra that do not match within 10 ppm to any peptide identified by MS/MS.
Small numbers of semitryptic peptides from abundant muscle proteins (like actin and myosin) were detected by MS/MS following LC separation, but they do not correspond to the unidentified peaks either. Most of these masses were also not mapped by PMF at any ranking to any protein substantiated by MS/MS identification. Together, these observations argue that there remain many peaks that can be readily observed in unseparated tryptic digests that cannot be explained yet.
The above discussion explains why PMF cannot yet possibly get all of the correct answers starting from tryptic digests of chicken muscle. The masses that can be reproducibly measured can nonetheless be used for purposes of classification, using unbiased methods like PCA analysis. As mentioned above, many of the masses that can be identified can be readily correlated with pathways that distinguish muscle fibers.
The limited data gathered here clearly delineate three different categories of muscle tissue in chickens. By studying additional samples, it ought to be possible to determine how many other additional categories of muscle may be distinguishable. Such data could determine which myosin heavy chains are expressed in specific muscles, to particular regions of specific muscles, or correlate to the age of the animal, or vary widely between individual animals [11]. In humans, most skeletal muscles contain a mixture of slow twitch and fast twitch muscle. In many diseases, there is a conversion to slow twitch muscle, and also increased expression of myosin heavy chains that are mostly expressed before or soon after birth. There are also specialized skeletal muscles that control motion in eyes and in swallowing. There is also more divergent muscle myosin genes expressed in cardiac tissue and in smooth muscle. A great deal of work has been done to characterized in detail the expression patterns at the RNA level in a small number of samples. Due to its simplicity, the approach described here ought to be suitable for characterization of large numbers of samples, and can be applied to many different tissues and organisms.
Conclusions
PMF is capable of rapidly identifying and providing semi-quantitative information on as many ten abundant proteins from unseparated trypsin digestion of tissues. It is possible to distinguish between isoforms of abundant proteins like myosin that are specific to distinct tissues. Therefore, we propose using PMF to classify tissue samples.
