Abstract
Purpose: Glioblastoma (GBM) is the most common form of brain tumor and has a uniformly poor prognosis. Development of prognostic biomarkers in easily accessible serum samples has the potential to improve the outcomes of patients with GBM through personalized therapy planning.
Material/Methods: In this study pre-treatment serum samples from 30 patients newly diagnosed with GBM were evaluated using a 40-protein multiplex ELISA platform. Analysis of potentially relevant gene targets using The Cancer Genome Atlas database was done using the Glioblastoma Bio Discovery Portal (GBM-BioDP). A ten-biomarker subgroup of clinically relevant molecules was selected using a functional grouping analysis of the 40 plex genes with two genes selected from each group on the basis of degree of variance, lack of co-linearity with other biomarkers and clinical interest. A Multivariate Cox proportional hazard approach was used to analyze the relationship between overall survival (OS), gene expression, and resection status as covariates.
Results: Thirty of 40 of the MSD molecules mapped to known genes within TCGA and separated the patient cohort into two main clusters centered predominantly around a grouping of classical and proneural versus the mesenchymal subtype as classified by Verhaak. Using the values for the 30 proteins in a prognostic index (PI) demonstrated that patients in the entire cohort with a PI below the median lived longer than those patients with a PI above the median (HR 1.8, p=0.001) even when stratified by both age and MGMT status. This finding was also consistent within each Verhaak subclass and highly significant (range p=0.0001-0.011).
Additionally, a subset of ten proteins including, CRP, SAA, VCAM1, VEGF, MDC, TNFA, IL7, IL8, IL10, IL16 were found to have prognostic value within the TCGA database and a positive correlation with overall survival in GBM patients who had received gross tumor resection followed by conventional radiation therapy and temozolomide treatment concurrent with the addition of valproic acid.
Conclusion: These findings demonstrate that proteomic approaches to the development of prognostic assays for treatment of GBM may hold potential clinical value.
Introduction
Glioblastoma (GBM) is the most common form of primary brain tumor found in adults in the United States. The current standard of care includes surgical resection, a concurrent course of radiation therapy (RT) and temozolomide (TMZ) treatment, followed by adjuvant TMZ administration with a median survival of 14.6 months [1]. However, even with this extremely poor prognosis, there is a small subset of patients that lives 5 years (10-15%) [2]. Thus, attempts to develop screening tools with prognostic value for the identification of those patients with better outcomes have been undertaken. Molecular sub typing of GBM tumors such as The Cancer Genome Atlas (TCGA) have identified four main subtypes of GBM; Pro-Neural, Neural, Classical and Mesenchymal though the survival difference between these four subgroups was similar [3]. Genomic/epigenetic analysis has identified O6-alkylguanine DNA alkyltransferase (MGMT) promoter methylation status as both a prognostic and predictive factor useful for tailoring GBM treatment regimens [4]. Though MGMT status is perhaps the most accepted biomarker for medical management of patients with glioblastoma, several other molecular markers include mutation of Epidermal Growth Factor Receptor (EGFR) and Isocitrate dehydrogenase (IDH) may have prognostic value [5]. In addition, Platelet-Derived Growth Factor Receptor A (PDGFRA), Tumor Protein p53 (TP53) and Circulating Tumor Cells (CTC) have also been identified as biomarkers with associated correlations of gene expression and patient outcome for patients with GBM tumors [6]. However, a lack of standardization in detection of these potential biomarkers has limited their usefulness.
Though these biomarkers may be promising, they require access to primary tumor tissue from a surgical resection for analysis. As surgical resection is standard of care for most patients with a GBM, there should be accessibility to tissue specimens but limitations in the quantity of specimen and standardization of the collection and processing of tissues remain significant obstacles. Additionally, surgical resection itself remains an important clinical variable affecting overall survival as positive correlations have been shown between extent of resection and both overall and progression free survival supporting the clinical benefit of gross total resection over sub-total resection or biopsy [7]. As tumor tissue is sometimes unavailable or of limited quantity, there is a need to develop biomarkers derived from non-tumor tissue.
Though no validated circulating biomarkers for GBM have been integrated into clinical practice, the use of circulating biomarkers for management of GBM remains an attractive approach due to accessibility, the ability to take serial samples and lower costs. Emerging circulating biomarkers for GBM have been identified in blood, urine and cerebrospinal fluid. These include identification of circulating tumor cells, cell-free circulating tumor DNA and miRNA and extracellular vesicles containing tumor derived nucleic acids [8]. Proteomic biomarker studies of GBM have also demonstrated protein signatures with preliminary prognostic value in patients. Comparison of proteomic signatures in the plasma of patients with GBM using mass spectrometry has shown that the plasma concentrations of proteins Leucine-rich alpha-2- glycoprotein 1 (LRG1), C-reactive protein (CRP) and Complement component 9 (C9) correlated with tumor size [9] and a cell-surface membrane protein signature of CD44, Vascular Cell Adhesion Molecule 1 (VCAM1), Heme Oxygenase (decycling) 1 (HMOX1) and transforming growth factor, beta-induced (BIGH3) secreted in the plasma was able to differentiate GBM patients from healthy controls [10]. Additionally, the use of non-tumor specific proteins as biomarkers for GBM include matrix associated proteins such as YKL-40, matrix metalloproteinases such as matrix metalloproteinase-2 (MMP2) and matrix Metallopeptidase (MMP9), acute phase proteins such as haptoglobin, cell lineage specific proteins related to the central nervous system such as Glial Fibrillary Acidic Protein (GFAP) and S100 calcium-binding protein B (S100B) and many other cytokines and growth factors including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (FGF2) and Transforming Growth Factor Beta 1 (TGFB1) [8,11]. In this study we used a multi-plex cytokine assay on samples collected prior to definitive chemoradiotherapy for patients with GBM compared to overall survival and progression free survival.
Methods and Materials
Protocol information
As reported previously, an open-label, Phase II study (NCI06-C-0112) was conducted at the National Cancer Institute and Virginia Commonwealth University in patients with histologically confirmed GBM, aged 18 years or older and a life expectancy greater than 8 weeks, with surgery no more than 6 weeks prior to enrollment [12]. The protocol was reviewed and approved by the NCI Institutional Review Board, and written informed consent was signed by all patients. The samples used in this study were collected from each patient prior to the initiation of therapy. Patients were then treated with daily valproic acid/irradiation/temozolomide for 6 weeks. Treatment response was analyzed per Response Evaluation Criteria in Solid Tumors (RECIST) and retrospectively by RANO criteria [13,14]. Time to progression was determined from initiation of treatment on protocol to symptomatic or radiographic progression. Overall Survival (OS) was determined from the initiation of treatment on protocol to date of death.
Multiplex biomarker assay
Patient serum was used for screening of protein biomarkers using the commercially available Mesoscale Diagnostics V-plex ELISA. This 40-plex assay included Pro-inflammatory Panel 1 (K15049D), Vascular Injury Panel 2 (K15198D), Angiogenesis Panel 1 (K15190D), Cytokine Panel 1 (K15050D), Chemokine Panel 1 (K15047D). This study included pre-treatment serum samples taken from 30 patients. The assays were run according to the manufacturer’s instructions.
Statistical methodology
Analysis of the potential biomarkers was performed using The Cancer Genome Atlas database and the Glioblastoma Bio Discovery Portal (GBM-BioDP), a freely accessible web resource that hosts a subset of the glioblastoma TCGA data and enables detailed queries and an interactive display of the resultant data [15]. Results from the multiplex assay were exported into R statistical programming language for further analyses [16]. The data was first normalized to z-scores, with the mean of the data adjusted to zero and the standard deviation adjusted to 1. The relative association in the strength of the features was assessed by pair wise correlations using the Pearson correlation method. The heat map analysis, used to visualize the correlation matrix, and the Kaplan-Meier plots, comparing the prognostic index with overall survival, were generated with the BioDP software.
To find potential multi collinearity among attributes, a matrix of pair wise correlations was built using the Pearson correlation method. The data was displayed as a correlation plot with positive and negative correlations displayed as blue or red color respectively. The intensity and size of the data points (circles) were made proportional to the correlation coefficients. The correlation coefficients were called significant at p ≤ 0.05, with the plot shown with only significant values.
A ten biomarker subgroup of clinically relevant molecules was selected using a functional grouping analysis of the 40 plex genes based on inflammatory, angiogenic, immune cell, chemotaxis or acute phase response characteristics. Two genes were selected from each of these functional groups based on the internal degree of variance, the lack of co-linearity with other biomarkers and clinical interest. Variance was calculated using the VARPA syntax in Excel.
A Multivariate Cox proportional hazard approach was used to analyze the relationship between overall survival (OS), gene expression, and resection status as covariates. A multi gene prognostic index is computed by weight averaging each gene’s expression by its regression coefficient in the Cox model [17]. Briefly, prognostic index (PI), also known as the risk score, is commonly used to generate risk groups. The PI is known as the linear component of the Cox model, PI= β 1 × 1+ β 2 × 2+…+ β pxp where xi is the expression value and the β I can be obtained from the Cox fitting. Each β I can be interpreted as a risk coefficient. The fitting is performed in rusing the “survival” package. The risk groups were dichotomized by ordered PI by median value (higher values for higher risk) leaving equal number of samples in each group. For the resulting two groups, Kaplan-Meier curves were then performed with expression values dichotomized according to the median value using R.
Results
Previously, Nijaguno et al. showed that a serum signature of eighteen cytokines could distinguish patients with a GBM from normal healthy volunteers [18]. To expand on this we used the Mesoscale Diagnostics 40-plex ELISA including a panel of chemokine, cytokine, angiogenic, vascular injury and pro inflammatory markers to determine if a cytokine signature could differentiate between short- and long-term survivors in patients diagnosed with a GBM [19]. The program BioDP was used to explore The Cancer Genome Atlas (TCGA) database to determine if these biomarkers were typically present in samples from patients with GBM [15].
As shown in the hierarchical cluster (HC) in (Figure 1A) 30 of 40 of the MSD biomarkers mapped to known genes within the TCGA and separated the 30 patients into two main clusters. The left cluster (red bar) had a higher number of samples, classified as classical and proneural, the right cluster (blue bar) had more samples classified as mesenchymal, and the neural subclass was distributed throughout [3]. The values for these 30 biomarkers were used to create a prognostic index (PI) that was compared to overall survival using Kaplan-Meier analysis. In the left most plot in (Figure 1B) patients in the entire cohort with a PI below the median lived longer than those patients with a PI above the median (HR 1.8, p=0.001), even when stratified by both age and MGMT status. Similarly, the patients with a PI below the median lived longer for each Verhaak subclass, which was highly statistically significant (range p=0.0001-0.011). The implication of this finding is that cytokine biomarkers could be used as a prognostic marker for patient survival in patients diagnosed with a GBM. Thirty of the thirtyseven patients enrolled on a Phase-2 study (NCI-06-C-0112) receiving VPA/RT/TMZ treatment had adequate amounts of stored sample and were used to validate the prognostic index. The clinical demographics of these patients are shown in table 1. The majority of patients were male, had a gross total resection and a Karnofsky performance score >90. Cytokine levels from samples collected from each patient, prior to the initiation of treatment, were processed using the MSD assay and representative values from four biomarkers are shown in figure 2. The data from the entire 40-plex biomarker panel are included in Supplemental table 1. In this VEGF-related grouping there is no significant finding for any one molecule. Importantly, the samples are graphed by order of accrual and we show that the time at -20°C storage does not introduce degradation of our biomarkers as both early and late collections had similar range of values. These data suggest that the biomarkers are stable over time at -20°C and that no individual protein can be used as a predicative biomarker.
| |
Characteristic |
n |
% |
| Age(y) |
54.3 (range: 31.8-71.5) |
|
|
| Sex |
Male |
22 |
73 |
| |
Female |
8 |
27 |
| RPA |
3 |
11 |
37 |
| |
4 |
12 |
40 |
| |
5 |
2 |
7 |
| |
Unknown/missing |
5 |
17 |
| Resection |
GTR |
16 |
53 |
| |
STR |
13 |
43 |
| |
Biopsy |
1 |
3 |
| KPS |
Median |
90 |
|
| |
Range |
80-100 |
|
| Tumor location |
Frontal Lobe |
10 |
|
| |
Temporal Lobe |
8 |
|
| |
Parietal Lobe |
7 |
|
| |
Occipital Lobe |
1 |
|
| |
Frontotemporal |
1 |
|
| |
Frontoparietal |
0 |
|
| |
Temporoparietal |
0 |
|
| |
Parieto-occipital |
3 |
|
| |
Temporo-occipital |
0 |
|
Table 1: Clinical Study Pretreatment Characteristics.
Clinical characteristics of patients included in the study cohort with RPA=Recursive Partitioning Analysis, GTR=Gross-Total Resection, STR=Sub-Total Resection, KPS=Karnofsky Performance Score.
Though we showed a 30-plex of data was predictive of overall survival in figure 1 using the 40-plex as a point of care assay for individual patients would be cost prohibitive. To reduce the number of biomarkers we evaluated the co-linearity between targets and eliminated biomarkers which had high co-line arities from further analysis. Correlated variables are shown in figure 3. Positive correlations are displayed in blue and negative correlations are shown in red. Color intensity and the size of the circle are proportional to the correlation coefficients. A p-value of (p>0.05) was called insignificant and empty cells represent insignificant pairs. To further reduce the number of targets, we organized the remaining molecules by class of function including inflammation, angiogenesis, immune cell, chemotaxis and acute phase response. Two biomarkers with a high internal variance were selected from each class for further multivariate analysis including: interleukin 7 (IL7), tumor necrosis factor alpha (TNFA), vascular cell adhesion protein 1 (VCAM1), vascular endothelial growth factor (VEGF), interleukin 10 (IL10), pro-interleukin-16 (IL16), interleukin 8 (IL8), C-C motif chemokine 22 (MDC), c- reactive protein (CRP), and serum amyloid A (SAA). This panel of ten biomarkers was then evaluated using the TCGA database to determine if this subgroup could also be predictive of clinical outcome for patients with GBM. We found that the 10-protein prognostic index was significant across all cohorts (p=0.0001-0.012) even when stratified by age and MGMT status, (Figure 4). These data indicate that using only the 10-protein panel maintained the predictive power of the larger panel when using the TCGA dataset.
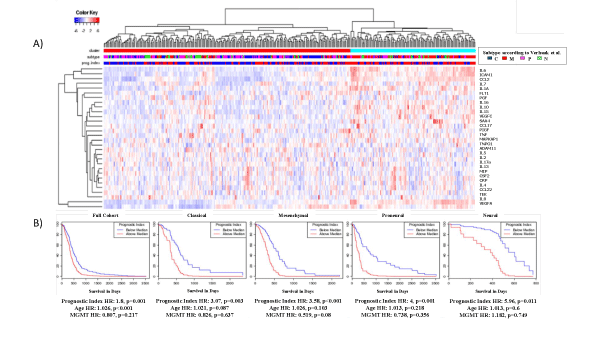
Figure 1: Analysis of 40 Biomarkers in TCGA.
A) Hierarchical clustering of TCGA GBM gene expression data showing 30 of the 40 biomarkers included in the Mesoscale Diagnostics V-plex ELISA platform. Subtypes: C=classical, M=mesenchymal, P=proneural, N=neural.
B) Kaplan-Meier plots based on a prognostic index generated from the selected 30 biomarker cohort showing the differences by using full cohort and Verhaak et al. [3]. Classified subclasses. Multivariate analysis included the prognostic index, age and MGMT status.
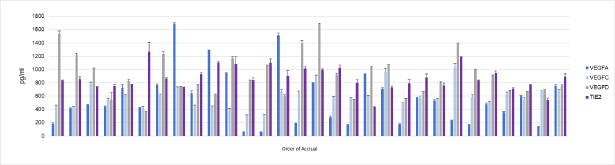
Figure 2: Selection of Biomarker Expression Values.
Serum expression of four biomarkers VEGFA, VEGFC, VEGFD and TIE2 randomly selected from amongst the 40 biomarkers included in the Mesoscale Diagnostics V-plex ELISA platform. Values are in pg/ml and samples are arranged by order of accrual.
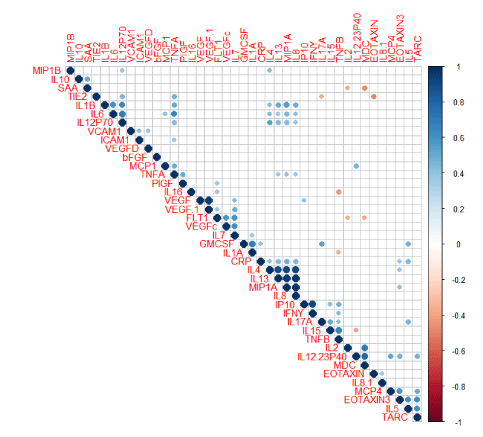
Figure 3: Analysis of Co-linearity of 40 Biomarkers.
Collinearity matrix of the 40 biomarkers tested by the ELISA platform. Positive and negative correlations are displayed as blue or red respectively. Darker shades of blue and red color indicate high variable collinearity while lighter shades indicate low collinearity. These intensity and size of the data points (circles) are proportional to the correlation coefficients. Correlation coefficients were called significant at (p ≤ 0.05), with the plot shown with only significant values.
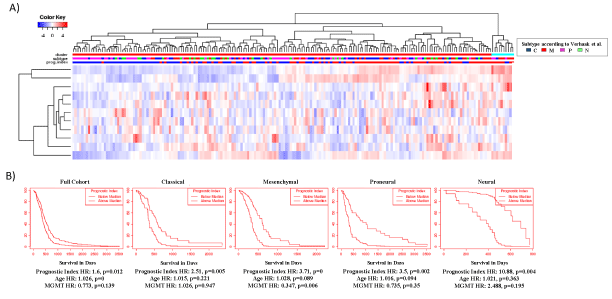
Figure 4: Analysis of 10 Biomarker Cohort in TCGA.
A) Heatmap showing hierarchical clustering of 10 biomarkers selected as described in the methods, represented by functional relevance. Subtypes: C=classical, M=mesenchymal, P=proneural, N=neural.
B) Kaplan-Meier plots based on a prognostic index generated from the 10 biomarkers are shown for full cohort and separated by Verhaak et al. [3] subclasses. Multivariate analysis included the prognostic index, age and MGMT status.
Next, we tested the 10-protein panel’s ability to predict overall survival using our 30 clinical specimens. As shown in figure 5A VCAM1, IL8 and IL16 had positive correlations with overall survival but none reached statistical significance. However, as shown in figure 5B when using a prognostic index derived from all 10 proteins the median split the group into two cohorts whose survival was different, though not statistically. The cohort that lived longer had values below the median suggestive of a less inflammatory/angiogenic tumor. Besides age and MGMT status, as shown previously, the third clinical feature that is predictive for clinical outcome is the amount of tumor resection. In our thirtypatient cohort 15 patients had a Gross Total Resection (GTR) and 14 had a Sub Total Resection (STR), one had a biopsy and was not further studied. As shown in figure 5C in patients that had a GTR the prognostic index split the patients into two cohorts with statistically significantly difference in survival. The patients whose PI was below the median survived the longest. This was not replicated in patients that had a STR, (Figure 5D). These data suggest that a panel of serum proteins may be useful in predicting future clinical outcome. This would need to be confirmed in a larger clinical cohort.
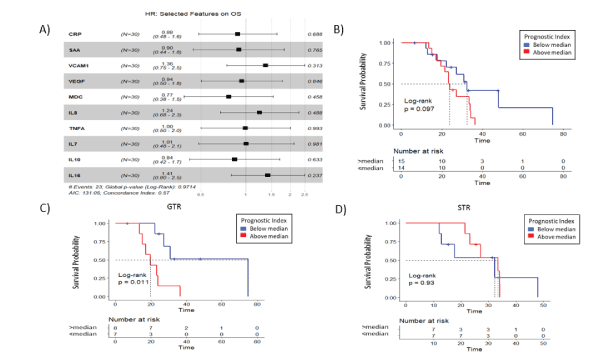
Figure 5: Verification of 10 Protein Biomarker Cohort Prognostic Index.
A) Cox proportional hazard was estimated for the 10 biomarkers using the current study GBM cohort. The plot shows the hazard ratios including confidence interval and p value. Kaplan- Meier plots using the 10 biomarker prognostic index for GBM patients were grouped by B) Overall Survival, and additionally by resection status C) Gross Tumor Resection (GTR) and D) Sub-Total Resection (STR).
Discussion and Conclusion
Due to the universally poor clinical outcome associated with Glioblastoma diagnosis, efforts to maximize treatment efficacy through personalized medicine have focused on development of biomarker signature assays with clinical potential. Identification of transcriptome signatures correlating with survival and genomic signatures which can distinguish between rapidly and slowly-progressing GBM using the TCGA database have identified promising approaches to develop new prognostic assays [20,21]. A recent study has also used the TCGA database to correlate consensus gene signatures with drug concordance to identify new preclinical drugs with potential for more effective GBM therapies [22]. In the current study, a panel of ten serum soluble protein biomarkers was found to be predictive of clinical outcome within the TCGA cohort and to correlate with overall survival in patients who underwent gross tumor resection and received conventional RT and concurrent TMZ treatment with the addition of VPA. Future studies should expand on these findings to test whether our ten protein biomarker panel was predictive of overall survival in GBM patients who received gross tumor resection and conventional RT and TMZ treatment in the absence of concurrent VPA administration.
Circulating biomarkers are needed to improve diagnosis and clinical assessment of GBM. Currently there are no such diagnostics available though there are recent clinical trials designed to address this confounder [23]. Though emerging prognostic molecular markers for GBM have been identified, they rely predominantly on tissue biopsy from the primary tumor [6]. Development of proteomic biomarkers secreted in the blood has advantages over tissue derived genomic and transcriptomic biomarkers due to ease of sampling. The current study demonstrates the utility of such an approach using a commercially available multiplex ELISA assay. Other recent studies have also focused on development of circulating biomarkers for GBM with potential diagnostic, prognostic or predictive value [8].
Biomarkers may also play an important role in the post-treatment setting. Radiation and systemic therapies can result in pseudo progression or radiation necrosis that can be difficult to distinguish from tumor growth. Currently no imaging tests can accurately differentiate these entities, with histological diagnosis remaining the gold-standard. However, re-resection is not possible in all patients due to tumor location or functional status. In those that are eligible, re- excision can be associated with significant morbidity, can provide inconclusive results due to sampling error and in many cases proves to be unnecessary. Biomarkers that can demonstrate tumor recurrence without invasive procedures would improve patient selection for surgery allow more accurate diagnosis in inaccessible tumors and limit the number of patients undergoing unnecessary operations. Our study demonstrates the feasibility of future proteomic biomarker-based assays to positively impact clinical assessment of GBM tumors.
Article Information
Article Type: RESEARCH ARTICLE
Citation: Sproull M, Mathen P, Miller CA, Mackey M, Cooley T, et al. (2019) A Serum Proteomic Signature Predicting Survival in Patients with Glioblastoma. J Biochem Analyt Stud 4(1): dx.doi.org/10.16966/2576-5833.117
Copyright: © 2019 Sproull M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
Received date: 01 Oct, 2019
Accepted date: 22 Oct, 2019
Published date: 26 Oct, 2019
