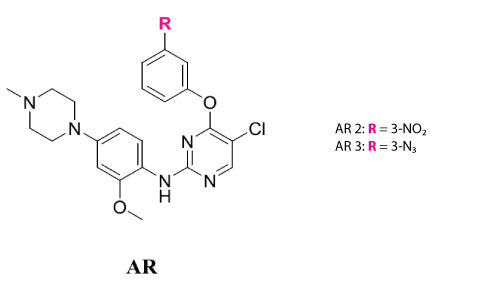
Figure 1: Structure of the anticancer drug candidate.

Aireen Romu1,2 Angie Li1 Karen Chen1 Vijaya Korlipara2 Enju Wang1*
1Department of Chemistry, St. John’s University, Jamaica, New York, USA*Corresponding author: Enju Wang, Department of Chemistry, St. John’s University, Jamaica, New York, USA Tel: (718) 990-5225; E-mail: wange@stjohns.edu
This work presents the studies of the interactions of two newly synthesized anticancer drug candidates (AR2 and AR3, analogues of WZ4002) with anilinopyrimidine scaffolding core moiety with bovine serum albumin (BSA) and human serum albumin (HSA) using UV-Vis absorption, fluorescence spectroscopy and molecular docking techniques. UV-Vis spectroscopy reveals an interaction of the two compounds with the albumins. Synchronous and regular fluorescence spectra of these two compounds show an exhibition of a static quenching process as formation of both BSA/HSA-drug complexes. The calculated binding constants with the correction of the absorptions of these compounds at 280 nm were moderately large. The calculated thermodynamic parameters (ΔH, ΔS and ΔG) reveal interaction forces between these two compounds and BSA/HSA fluorescence sites (reside in site I sub-domain IIA) are through Van der Waals (hydrophobic) and hydrogen binding forces which was also observed in the molecular docking study.
UV-Vis; Fluorescence spectroscopy; Molecular docking; BSA and HAS binding; anilinopyrimidine; EGFR-TKIs
Serum albumin (SA) such as bovine (BSA) and human (HSA) is the most abundant blood plasma protein acting as the transporter and carrier of many endogenous (fatty acids, amino acids and ions, etc.) and exogenous (drugs) substances [1,2]. It is a well-established finding that albumin’s binding ability to a variety of drugs impact their rate of delivery, efficacy and alteration of drug structural properties, which is a subject of great interest in the pharmaceutical industry [3,4]. Drug protein binding studies can provide important information to understand the pharmacodynamics and pharmacokinetics of the drugs, and also the structure and function of the protein [5-7]. The binding nature and magnitude of drug-protein, specifically drugs with SA, affect pharmacological behavior, ADMET (adsorption, distribution, metabolism, elimination and toxicity). Strong binding affinity of the drug-SA complex decreases free drug concentration, reduces toxicity, increases solubility of hydrophobic drugs in blood plasma and modulates their delivery to cells. Whereas, weaker binding shortens the life expectancy of the drug and leads to its poor distribution to the target tissue [8,9]. As a result, in vitro binding studies of drug interaction with SA are very important and give insights for designing better drugs for pharmacological activity and efficacy [9].
SA is widely used as a model protein for the investigation of protein surface binding and drug binding mechanisms because of its unique microenvironment (~seven hydrophobic grooves on its surface), and because it acts as a universal receptor for many drug molecules [10,11]. Both BSA and HSA are studied extensively side by side for protein-protein or protein-drug interaction because of their 76% similar structural sequence homology (583 and 585 amino acid residues respectively) [12]. They both are a single chain globular protein containing three structurally homologues domains, I, II and III along with two sub-domains (A and B) within each. Two specific drug binding sites were identified in the hydrophobic cavities in the sub-domains, known as sites I (in subdomain IIA) and II (in subdomain IIIA) of albumin [13-15]. Protein fluorescence originates from amino acid residues such as tryptophan, tyrosine and phenylalanine present at these sites or from the fluorescence of the bound drug molecule itself. In BSA, the two dominant fluorescence sites are Trp 134 located on the surface of sub-domain IB, and Trp 212 located within the hydrophobic pocket of sub-domain IIA. The amino acids responsible for its intrinsic fluorescence in HSA are Trp 214 (most dominant) which resides in sub-domain IIA, and several tyrosine residues located in different sub-domains [14-16].
Fluorescence spectroscopy is a commonly used analytical technique in the study of interactions between drugs and serum albumin because of its high sensitivity, selectivity and susceptibility to the local chemical environment of the fluorescence group [17,18]. Solvent and ligands in the solution could induce the protein conformational change and reduce the emission through dynamic and static quenching [17]. It is through fluorescence quenching measurements, one can acquire drugalbumin binding information. Depending on the chemical properties of the compounds, e.g. size, polarity, charge and UV/Vis absorption ability, the fluorescence quenching ability, the binding strength (with binding constant), the accessibility (the number of binding sites) and the binding forces to the two Tryptophan sites are significantly different [18-20].
The goal of this project is to investigate the binding affinity of two anticancer drug candidates (AR2 and AR3) with anilinopyrimidine scaffolding core moiety containing electrophilic groups that were synthesized as epidermal growth factor receptor (EGFR) inhibitors (Figure 1). Pyrimidine moiety is one of the important classes of N-containing heterocycles molecules broadly used as key building blocks for pharmaceutical agents, specifically as anticancer agents [21,22]. AR2 and AR3 with NO2 or N3 as substituting groups, were derived from WZ4002 (a third-generation of EGFR-TKIs), which is an irreversible inhibitor designed to treat non-small cell lung cancer patients harboring T790M mutation [23]. Cytotoxicity assay studies have been done on AR2 and AR3 against different cell lines (PC9, PC9-GR, H460, Ba/f3WT and Ba/f3T315I) in comparison to gefitinib and imatinib. Both AR2 and AR3 were found to exhibit similar or better potency against PC9-GR and H460 cell lines in comparison to gefitinib. They also exhibited better potency against Ba/F3 T3151 in comparison to imatinib. In comparison to WZ4002, they have similar or lower IC50 value against PC9-GR, H460, Ba/F3 WT and Ba/F3T3151 cell lines [23]. As a result, studying the interaction of these two compounds with SA has biological relevance. The studies were done using UV-Vis, fluorescence spectroscopy and molecular modeling to understand the carrier role of serum albumin for such compounds in the blood plasma under physiological conditions.
Figure 1: Structure of the anticancer drug candidate.
Commercially prepared bovine serum albumin (BSA, purity 98.0%) and human serum albumin (HSA, purity 98.0%) were purchased from Sigma-Aldrich Co. (USA) and stored in refrigerator at 4.0°C. The stock solutions: 2.1 × 10-3 M of AR2 and AR3 were prepared in methanol and 1.00 × 10-5 M BSA/HAS were prepared in 0.10 M phosphate buffer (PBS, pH 7.2). The BSA/HSA stock solutions was then stored in the dark at 4.0°C before fluorescence and UV-Vis absorption assay. All other reagents of analytical grade and were used as purchased without further purification. Double distilled and de-ionized water was used for the preparation of all aqueous solutions.
Compounds AR2 and AR3 were synthesized according to the procedures as described in our previous reports [19].
UV-Vis Absorption Measurements: The absorption spectra were recorded on a Thermo Insight UV-Vis spectrophotometer evolution 220, using a 1.0 cm quartz cell. The UV-Vis absorption spectra of AR2/ AR3 solutions in the concentration range of (1.76-35) × 10-6 M, BSA/ HSA of (5-10) × 10-6 M, and the mixture of BSA-AR compound in various concentration ratios in a pH 7.2 phosphate buffer at 298 K were recorded in the range of 250 to 400 nm.
Fluorescence Measurements: All fluorescence regular and synchronous spectra were obtained on a Perkin Elmer LS50B spectrophotometer equipped with a thermostatic bath using a 1.0 cm quartz cuvette. The fluorescence measurements were performed at three temperatures (293, 298, 303 K) in the range of 290-500 nm. For the fluorescence spectra measurements, 100 µL aliquot of 1.00 × 10-5 M BSA/HSA solution was added to 2900 µL of buffer solution. The BSA/ HSA solution was titrated by successive additions of stock compound solutions manually, and the AR2/AR3 concentration varied from 0 to 20 µM. The excitation and emission slit widths were both set at 10 nm. The excitation wavelength was 280 nm and the temperature of samples was maintained by recycling water during the whole experiment.
Synchronous spectra were obtained from 270-350 nm at Δλ=15 nm and Δλ=60 nm with the excitation and emission slits set at 10 nm.
Molecular Modeling: All docking in-silico studies experiment were performed on a Mac Pro 6-core Intel Xenon X5 processor with Macintosh Operating System (OS X El Capitan) using the Schrodinger 20175-1 (Schrӧdinger, LLC, New York, NY, 20175) software. Ligand preparations (LigPrep, Schrӧdinger, LLC, New York, NY, 2017) of the anilinopyrimimidine compounds was performed using default parameters, homology modeling (except using template as PDB: 4F5S and 1E7H), AR2 and AR3 and protein preparation of the homology model were essentially performed following reported protocols [24,25]. Protein preparation wizard tool (Schrӧdinger Suite 2017- 1 Protein Preparation Wizard; Epik, Schrӧdinger, LLC, New York, NY, 2016; Impact, Schrӧdinger, LLC, New York, NY, 2016; Prime, Schrӧdinger, LLC, New York, NY, 2017.) was employed to refine the structures of Bovine Serum Albumin (BSA) (PDB: 4F5S) and Human Serum Albumin (HSA) (PDB: 1E7H). Grid preparation was performed by using the centroid of amino acid residue Tyr 149 and Trp 214 for BSA and HSA protein respectively. Docking (Glide, Schrӧdinger, LLC, New York, NY, 2017) was performed using Standard Precision (SP) method using default parameters. Homology model validation was performed based on Ramachandran plot and Rootmean-Square Deviation (RMSD) of Cα atoms of the residues of the generated model and experimental structure. The Ramachandran plot analysis revealed >94% residues in the favored region, ∼4% residues in the allowed regions, and ∼1.2% residues (all glycine and Ala82) in the disallowed regions. The RMSD was found to be 0.061 Å, suggesting good alignment. The grid was generated by selecting residues at 4 Å from bound inhibitors in homology modeled proteins (template PDBs: 4F5S and 1E7H). The default protocol was used to perform induced-fit docking except the number of poses was reduced to 10. The surface representation of the bound ligand is generated by using the “create binding site surfaces” tool in Maestro with default parameters and a residue-type color scheme.
UV-Vis spectra studies provide insight about the structural changes and protein-substrate complex formation. The two pyrimidine analogues UV-Vis spectral analysis were done in the presence and absence of both BSA and HSA. BSA and HSA absorbs in the UV range below 250 nm to 300 nm with the λ max around 280 nm. AR2 and AR3 have an aromatic ring with a broad absorption peak at 270 nm. There is an overlap of absorption spectra of BSA and AR compounds at 280 nm. Figure 2 shows absorbance spectra of 7.0 × 10-6 M BSA with AR3. With both AR2 and AR3, the absorbance of the individual pyrimidine compound is slightly lower than the additive of BSA with each pyrimidine compound absorbance of the same concentration. At higher concentration of the AR compounds these differences of absorbance spectra curve 3 and 4 are smaller. Similar findings were observed with HSA UV-Vis absorption spectra changes as well with the two AR compounds. These results indicate that there is an interaction between BSA/HSA and the pyrimidine compounds which altered the electronic energy levels of both compounds slightly. However, substitution on the phenoxy ring with -N3 or -NO2 did not show a significant difference.
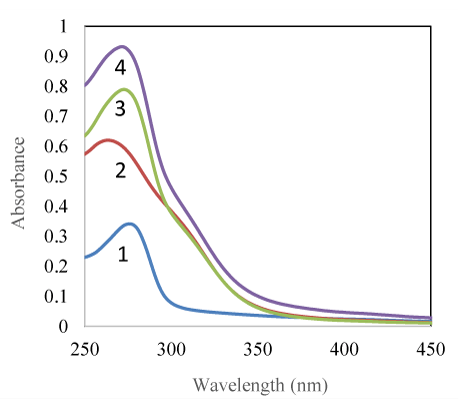
Figure 2: Absorbance spectra of (1) 7.0 × 10-6 M BSA, (2) 4.3 × 10-5 M AR3, (3) 7.0 × 10-6 M) BSA and 4.3 × 10-5 M AR3, and (4) the additive of (1) and (2), all at pH 7.2.
The micro-environment around the fluorophore consists of three amino acids residues, tyrosine, tryptophan and phenylalanine. Change of fluorescence is observed when these three amino acids are altered due to any structural or conformational change of the protein at the binding site. To understand the binding interaction mechanism of the protein-ligand complex formation at the cavity, fluorescence experiments of the drug candidates (AR2 & AR3) were performed in both BSA and HSA. The fluorescence data for both proteins were collected and evaluated. Due to similarity, only fluorescence spectra of BSA at pH 7.2 with addition of compound AR3 at different concentrations are presented as an example in figure 3. Both BSA and HSA exhibited a strong fluorescence emission peak at 345 nm when excited at 280 nm. A decrease in the fluorescence intensity of the protein at 345 nm peak was observed as the concentration of AR2/AR3 was gradually increased to 1.5 × 10-5 M. No observable shifts of the emission peak were observed. These results show that the fluorescence sites are experiencing a micro-environment change with the addition of the AR compounds.

Figure 3: Fluorescence spectra of 3.3 × 10-7 M BSA in absence and presence of AR3 at pH 7.2 and 298 K. AR3 concentrations from top to bottom: 0, 1.7, 3.5, 5.2, 7.0, 8.7, 10.4, 12 and 14 (× 10-6M).
Synchronous fluorescence spectra were employed to investigate the protein conformational changes. If the Δλ between the excitation wavelength and emission wavelength is set at 15 nm and 60 nm, the synchronous fluorescence spectra provide the characteristic information of tyrosine or tryptophan residues respectively [26,27]. Figure 4ab showed that their emission intensities decreased with increasing concentration of AR3, in addition, the fluorescence intensity of tryptophan is much stronger than that of tyrosine although the quenching by AR3 is to a similar degree. From figure 4a (Δλ=15 nm) and 4b (Δλ=60 nm), there are no shifts in both the emission wavelengths of both tyrosine and tryptophan residues.
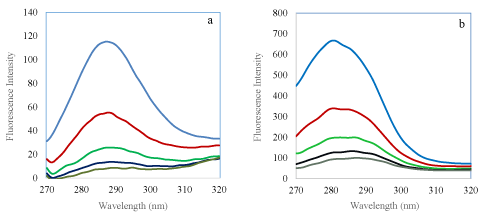
Figure 4: Synchronous fluorescence spectra of 3.3 × 10-7 M BSA in the absence and in the presence of AR3 (a) at ∆λ=15 nm and (b) ∆λ =60 nm at 298 K and pH 7.2. AR3 concentrations from top to bottom: 0, 3.5, 7.0, 12, 14 (× 10-6 M).
Fluorescence quenching results from a variety of molecular interactions (excited-state reactions, energy transfer, ground state complex formation, molecular rearrangements and collisional quenching) to decrease the fluorescence intensity of the protein-ligand complex [28]. To study the quenching mechanism (dynamic or static quenching), the fluorescence measurements were analyzed using the Stern-Volmer equation:
\[\frac{{{F_0}}}{F} = 1 + {K_{sv}}\left[ Q \right] = 1 + {k_q}{t_0}\left[ Q \right]\,\,\,\,\,\,\left( 1 \right)\]
Where F0 (absence of quencher) and F (presence of quencher) are the fluorescence intensities of the drug molecules. kq is the quencher rate constant, τ0 is the average life time of the emissive excited state of fluorescence molecular without the presence of the quencher and [Q] is the concentration of the quencher. Ksv is the Stern-Volmer dynamic quenching constant. kq can be calculated from Ksv in biomolecule when τ0 is known: kq =Ksv/τ0 .
Since there is absorption at 280 nm by AR2 and AR3, the fluorescence was corrected [26,27,29] with the equation:
\[Fcor = F{e^{A/2}}\,\,\,\,\,(2)\]
Here, F is the fluorescence intensity measured at the corresponding concentration of the quencher; A is the absorbance of the quencher at 280 nm with A less than 0.3. Equation (1) is modified to:
\[\frac{{{F_0}}}{{{F_{cor}}}} = 1 + {K_{sv}}\left[ Q \right] = 1 + {k_q}{t_0}\left[ Q \right]\,\,\,\,\,\,\,\,(3)\]
Both quenching mechanisms are temperature dependent, at higher temperature the rate of diffusion is faster and the amounts of collisions are greater resulting in an increase in the dynamic quenching constant value; whereas, the opposite effect would be observed for static quenching (lower constant value) due to destabilization of weakly bound protein-drug complexes will occur at higher temperature. The fluorescence quenching of both serum albumins by the two compounds (AR2 & AR3) was evaluated and analyzed at three different temperatures (293, 298 & 303 K). The concentration of the AR compounds was kept below 2 × 10-5 M to ensure their absorbance below 0.3. The results are plotted and an example is shown in figure 5 with AR3. The Stern-Volmer plots exhibit a linear relationship of F0/Fcor vs the concentration of the two compounds AR2 and AR3 with both BSA and HSA. From these plots Ksv and Kq values were calculated and listed in table 1. The observed values of Kq (the rate constant of the quenching process) were 100 times greater than the maximum scatter quenching constant (2 × 1010 M-1s-1) of the biomolecule [30] indicating a static quenching mechanism exists for both the compounds in both serum albumins. The calculated Ksv values for AR2 in both BSA and HAS does not show a clear trend with increase in temperature. However, the Ksv value for AR3 to BSA/HAS quenching increases along with the increase in temperature. These indicate that both compounds exhibit a combination of dynamic and static quenching mechanism to both albumins [31,32].
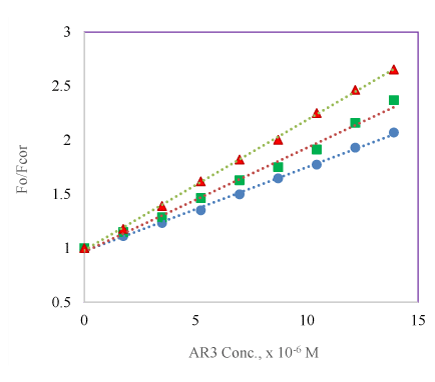
Figure 5: Stern-Volmer plot of the BSA (3.3 × 10-7 M, pH 7.2) with corrected fluorescence emission intensity at 345 nm in the presence of AR3 at ( ) 293 K, ( ) 298 K and ( ) 303 K.
| BSA | HSA | ||||
| Comp | T (K) | Ksv (104 M-1) | Kq (1012 M-1s-1) | Ksv (104M-1) | Kq (1012 M-1s-1) |
| AR2 | 293 | 8.29 | 8.29 | 6.37 | 6.37 |
| 298 | 8.8 | 8.8 | 7.06 | 7.06 | |
| 303 | 7.68 | 7.68 | 6.62 | 6.62 | |
| AR3 | 293 | 7.49 | 7.49 | 6.94 | 6.94 |
| 298 | 8.79 | 8.79 | 8.67 | 8.67 | |
| 303 | 11.9 | 11.9 | 8.66 | 8.66 | |
Table 1: The parameters of Stern-Volmer plots for the quenching of BSA and HSA by AR compounds at three different temperatures.
To further elucidate the static quenching mechanism of the interaction, the association constant (Ka) and number of binding sites (n) at various temperatures were determined using a modified SternVolmer equation:
\[\log \left[ {\frac{{{F_0} - {F_{cor}}}}{{{F_{cor}}}}} \right] = \log {K_a} + n\log Q\,\,\,\,\,\,\,(4)\]
Where Ka and n represents the binding constant and the number of binding sites respectively. All the other parameters represent the same as in the prior Stern-Volmer equation (Equations 1-3). The calculated Ka and the n values from the logarithm plot (see figure 6 for AR3) at various temperatures are presented in table 2. It is observed that as the temperature increased the Ka value also increased for AR3 in BSA and HSA, but for AR2 the Ka value seemed to be decreasing slightly with increase in temperature. A Ka value of 104 -105 order indicates that a moderate strong binding force is formed between each AR compound and BSA. AR2 has dominant static binding force with both BSA and HSA since the Ka value decreased with increase in temperature. In HSA, both drug candidate compounds have Ka values slightly smaller than with BSA. The n value of the entire protein-drug complex in both BSA and HSA at various temperatures is approximately 1, indicating the presence of only one binding site during interaction of the drug candidate molecules in both serum albumins. The drug compounds are likely bound to the tryptophan residue near or within the hydrophobic pocket located in the subdomain IIA [31,32].
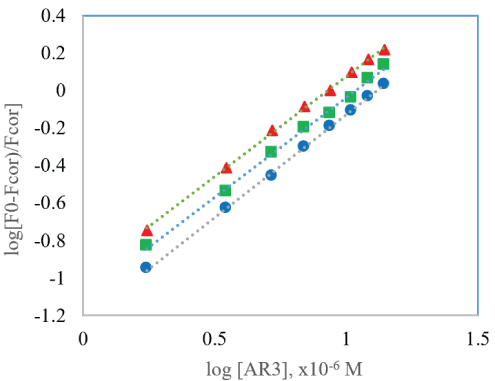
Figure 6: Modified Stern-Volmer plot of BSA-AR3 system at ( ) 293 K, ( ) 298 K and ( ) 303 K.
| BSA | HSA | ||||||||||
| Comp | T (K) | Ka × 104 (M-1) | n | ΔG (kJ/mol) |
ΔH (kJ/mol) |
ΔS (J/mol) |
Ka × 104 (M-1) | n | ΔG kJ/mol |
ΔH kJ/mol |
ΔS J/mol |
| AR2 | 293 | 11.9 | 0.88 | -28.5 | 4.1 | 109.2 | 9.11 | 0.85 | -27.8 | -4.1 | 79.5 |
| 298 | 9.39 | 0.98 | -28.4 | 7.84 | 0.96 | -27.9 | |||||
| 303 | 7.33 | 1.02 | -28.2 | 7.4 | 0.96 | -28.2 | |||||
| AR3 | 293 | 5.97 | 1.09 | -26.8 | -51.2 | -82.0 | 5.75 | 1.09 | -26.7 | -36.3 | -32.4 |
| 298 | 8.3 | 1.02 | -28.1 | 6.89 | 1.11 | -27.6 | |||||
| 303 | 16.8 | 0.88 | -30.3 | 8.2 | 1.03 | -28.5 | |||||
Table 2: The binding constants K, the number of binding sites n, and the thermodynamic parameters of BSA and HSA at different temperature.
There are four types of force/interactions (hydrogen bonding, hydrophobic, Van der Waals and electrostatic) identified between drugs and biological macromolecules. Each type of these forces can be determined by the signs and magnitudes of the thermodynamic parameters [enthalpy change (ΔH) and entropy change (ΔS)]: hydrophobic forces (+ΔH and +ΔS), hydrogen bonding and Van der Waals forces (-ΔH and -ΔS) and electrostatic forces (-ΔH and +ΔS) [32]. The spontaneity of the protein-drug interaction can be obtained from the sign and magnitude of free energy change (ΔG). Spontaneous interaction occurs at a -ΔG value, whereas nonspontaneous will be at a +ΔG. These three thermodynamic parameters can be calculated using the following equations:
ΔG=-RTlnK (5)
\[1n\frac{{{K_2}}}{{{K_1}}} = \frac{{\Delta H}}{R}\left[ {\frac{1}{{{T_1}}} - \frac{1}{{{T_2}}}} \right]\,\,\,\,\,\,\,\,\,\,\,(6)\]
∆G=ΔH–TΔS (7)
The calculated values of each parameter at various temperatures are listed in table 2. Accordingly, ΔG values for the two compounds are observed to be negative in both BSA and in HSA indicating a spontaneous binding formation. AR2 binds with BSA through hydrophobic interactions (both ΔH and ΔS are positive) at the binding site, whereas in HSA, it is through electrostatic interactions (-ΔH and +ΔS). AR3 interacts at the binding site with both serum albumin through hydrogen bonding and Van der Waals forces (-ΔH and -ΔS).
Molecular modeling
Molecular docking provides insight about the binding site pocket where protein-ligand complex bounds. It predicts the interactions between biomolecules and small molecules indicating the thermodynamic feasibility of the binding site region. Figure 7 illustrates the docking pose of the most favorable binding conformation for both compound AR2 and AR3 interacting with the residues of HSA and the residues of BSA, and the types of interactions involved. Based on the docking results depicted in table 3, sub-domain IIA (site I) of both SA is the most favorable binding site for our compounds.
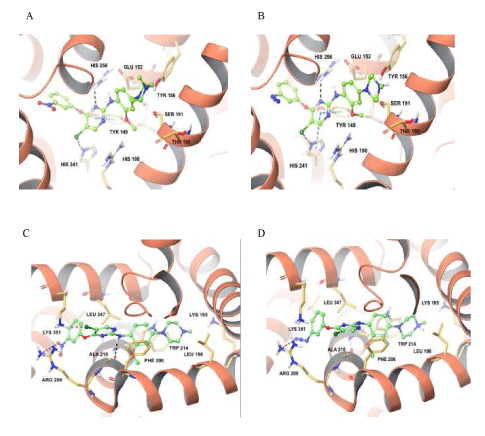
Figure 7: Induced-fit docking model of AR2 and AR3 within transmembrane domains of homology modeled serum albumin. (A) Docking pose of compound AR2 in BSA. Amino acid residues are illustrated as thin tubes with the color representations as carbon in faded orange, hydrogen in white, nitrogen in blue, oxygen in red, and chlorine in dark green. The protein is represented in the faded red orange colored ribbon form. The ball and stick model of molecule with the identical color scheme as above except carbon atoms represented in green. Black dashes represent proteinligand intermolecular hydrogen bonds and light blue dashes represent protein-ligand intermolecular π-π staking interactions. (B) Docking pose of compound AR3 in BSA. Representation is the same as in A. (C) Docking pose of compound AR2 in HSA. Representation is the same as in A and red dashes represent protein-ligand intermolecular electrostatic interactions. (D) Docking pose of AR3 in HSA. Representation is the same as in C.
| AR2 | AR3 | ||||
| Binding Site | Docking Score (kJ/mol) |
ΔGo (kJ/mol) |
Docking Score (kJ/mol) |
ΔGo (kJ/mol) |
|
| BSA | Site I | -23.1 | -28.4 | -27.3 | -27.9 |
| Site II | -12.9 | -18.8 | |||
| HSA | Site I | -35.6 | -28.1 | -32.3 | -27.6 |
| Site II | -19.2 | -18.0 | |||
Table 3: Docking score and Gibbs free energy of compounds binding to BSA and HSA at 298K.
In BSA, both drug candidates reside in a hydrophobic pocket slightly away from Trp 212 near two tyrosine residues, Tyr 149 and Tyr 156, and surrounded by other amino acids such as His 256, Glu 152, Ser 191, Thr 190, His 198 and His 24. The key interactions identified at the binding site region are the following: N3 on the pyrimidine ring of both AR2 and AR3 forms a H-bond with one of the -NH2 group of His 256. A π-π stacking interaction was observed between the pyrimidine ring of both AR2 & AR3 and the benzene ring of Tyr 149. In addition, for AR3 a second π-π staking interaction was found between the pyrimidine ring and the imidazole ring of His 241.
In HSA, the drug candidates are residing in a hydrophobic pocket near the Trp 214 residue and surrounded by the following amino acid residues: Lys 351, Leu 347, Lys 195, Leu 198, Phe 206, Ala 210 and Arg 209. The docking data displays the following key interactions at the binding site region of HSA: The -NH group of anilinopyrimidine ring of AR2 forms H-bonding interactions with the carbonyl (C=O) group of Ala 210. A π-cation interaction was found among the phenoxy ring of AR3 with Lys 351. The nitro group on AR2 and azido group on AR3 forms electrostatic interaction with Arg 209 side chain.
The theoretical binding energy obtained from molecular modeling was calculated and compared with the free energy (ΔG) spectroscopic values (Table 3). The results are comparable. The error does fall in the standard minimum binding energy error range (~8.36-12.54 kJ/mol)5.
The negative ΔG value for each compound indicates that the binding process is thermodynamically favorable at the binding site. In addition, the molecular docking information stipulates that hydrogen bonding and hydrophobic interactions play a major role for both compounds binding with BSA and HSA.
In summary, the binding interactions of AR2 and AR3 with serum albumin were investigated using various spectroscopic techniques (UV-Vis and fluorescence) along with molecular docking method. Both experimental and molecular docking results signified a thermodynamically favorable (ΔG<0) binding process at the binding sites of both BSA and HAS with each AR compound. The more negative the ΔG value, the higher the binding constant with the protein. According to the molecular docking, the preferred binding site for the AR compounds was at site I in both BSA and in HSA. The mechanism of the binding process was found to be a static quenching rather than a dynamic quenching mechanism based from the fluorescence data obtained. The two main types of interactions identified from the thermodynamic parameters (ΔH and ΔS) results at the binding site of BSA and HSA with the AR compounds are hydrogen bonding and hydrophobic interactions, which is also consistent with the molecular docking results. These important findings at the binding site of BSA and HSA will be useful in the pharmaceutical industry. It will provide critical insights and give a better understanding of drug protein binding relationships and new drugs discovery specifically for N-containing heterocycles molecules.
The authors declare that there are no conflicts of interest.
The authors thank Dr. Uday K. Velagapudi and Dr. Tanaji T. Talele, Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John’s University for molecular docking program access. Support for this research was provided by Saint John’s University. EW thanks the writing center of St. John’s University in facilitating the completion of the manuscript.
Download Provisional PDF Here
Article Type: RESEARCH ARTICLE
Citation: Romu A, Li A, Chen K, Korlipara V, Wang E (2019) UV-Vis, Fluorescence and Molecular Docking Studies on the Binding of Bovine and Human Serum Albumins with Novel Anticancer Drug Candidates. J Biochem Analyt Stud 4(1): dx.doi.org/10.16966/2576-5833.116
Copyright: © 2019 Romu A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access