Introduction
Alzheimer’s Disease (AD) is a polygenic/complex disorder in which
hundreds of polymorphic variants distributed across the human genome
are potentially involved, in conjunction with epigenetic phenomena,
cerebrovascular disorders and environmental factors, leading to
premature neuronal death and concomitant cognitive decline [1-4].
AD shares pathogenic features with conformational disorders in which
the abnormal expression of genes generates conformational changes
in key proteins (Amyloid beta (Aβ), hyperphosphorylation of MAPTTau),
contributing to the formation of senile plaques and neurofibrillary
tangles [1]. The pharmacological treatment of AD with conventional
drugs (donepezil, rivastigmine, galantamine, memantine) is not costeffective
and many novel therapeutic strategies are under development
worldwide [3]. Furthermore, AD patients may take 6-12 different
drugs/day for the treatment of dementia-related symptoms, including
memory decline (conventional anti-dementia drugs, neuroprotectants),
behavioral changes (antidepressants, neuroleptics, sedatives, hypnotics),
and functional decline, or for the treatment of concomitant pathologies
(epilepsy, cardiovascular and cerebrovascular disorders, parkinsonism,
hypertension, dyslipidemia, anemia, arthrosis, etc). Over 20% of
dementia patients are current users of cardiovascular drugs. A high
throughput screening study assessed 1,600 FDA-approved drugs for their
ability to modulate Aβ activity; 559 drugs of the 1,600 had no effect on
amyloid precursor protein (APP) processing or were toxic to neurons at
the testing concentration, while 800 drugs could reduce Aβ content by
over 10% in primary neurons derived from Tg2576 mice, among which
184 drugs were able to reduce Aβ content by over 30%; 241 drugs could
potentially promote Aβ accumulation, including 26 drugs that could
increase the level of Aβ by more than 30% [5]. The co-administration of
several drugs may cause side-effects and adverse drug reactions in over
60% of AD patients, who in 2-10% of the cases require hospitalization.
The assessment of the prevalence of Potentially Inappropriate Medication
(PIM) in French patients with mild-to-moderate AD showed that 46.8%
of the patients had at least one PIM [6]. “Cerebral vasodilators” were the
most widely-used class of PIM, accounting for 24.0% of all prescriptions,
followed by atropinic drugs and long half-life benzodiazepines. Atropinic
drugs were associated with cholinesterase inhibitors in 16% of patients.
In over 20% of the patients, behavioral deterioration and psychomotor
function can be severely altered by polypharmacy. The principal causes
of these iatrogenic effects are (i) the inappropriate combination of drugs,
and (ii) the genomic background of the patient, responsible for his/her
pharmacogenomic outcome.
Pharmacogenomics accounts for 30-90% variability in pharmacokinetics
and pharmacodynamics. The pharmacogenetic outcome is the result
of multiple gene interactions and their respective gene products
potentially involved in the therapeutic effect and/or toxicity of drugs
[3]. In addition, drug-drug interactions, concomitant pathologies,
and epigenetic changes in genes linked to the pharmacogenetic network
associated with efficacy and safety issues of a particular drug also affect
the final pharmacogenetic outcome [2-4].
The genes involved in the pharmacogenomic response to drugs in AD
fall into five major categories: (i) genes associated with AD pathogenesis
and neurodegeneration (APP, PSEN1, PSEN2, MAPT, PRNP, APOE
and others); (ii) genes associated with the mechanism of action of drugs
(enzymes, receptors, transmitters, messengers); (iii) genes associated with
drug metabolism (phase I (CYPs) and phase II reactions (UGTs, NATs); (iv)
genes associated with drug transporters (ABCs, SLCs); and (v) pleiotropic
genes involved in multifaceted cascades and metabolic reactions (APOs,
ILs, MTHFR, ACE, AGT, NOS, etc) [7,8]
Pathogenic Genes
Te genetic and epigenetic defects identified so far in AD include
Mendelian mutations, susceptibility Single-Nucleotide Polymorphisms
(SNPs), mitochondrial DNA (mtDNA) mutations, and epigenetic
changes. Mendelian mutations affect genes directly linked to AD,
including mutations in the APP gene (21q21) (AD1), mutations in
the presenilin 1 (PSEN1) gene (14q24.3) (AD3), and mutations in the
presenilin 2 (PSEN2) gene (1q31-q42) (AD4) [1, 9-13]. PSEN1 and
PSEN2 are important determinants of γ-secretase activity responsible
for the proteolytic cleavage of APP and NOTCH receptor proteins.
Mendelian mutations are very rare in AD (1:1,000). Mutations in exons
16 and 17 of the APP gene appear with a frequency of 0.30% and 0.78%,
respectively, in AD patients. Likewise, PSEN1, PSEN2, and MicrotubuleAssociated
Protein Tau (MAPT) (17q21.1) mutations are present in less
than 2% of the cases. In the Alzgene database [9] there are over 600 genes
potentially associated with AD, of which the top ten are APOE (19q13.2),
BIN1 (2q14), CLU (8p21-p12), ABCA7 (19p13.3), CR1 (1q32), PICALM
(11q14), MS4A6A (11q12.1), CD33 (19q13.3), MS4A4E (11q12.2), and
CD2AP (6p12). Potentially defective genes associated with AD represent
about 1.39% (35,252.69 Kb) of the human genome, which is integrated
by 36,505 genes (3,095,677.41 Kb). The highest number of AD-related
defective genes concentrate on chromosomes 10 (5.41%; 7,337.83 Kb), 21
(4.76%; 2,289,15 Kb), 7 (1.62%; 2,584.26 Kb), 2 (1.56%; 3,799.67 Kb), 19
(1.45%; 854.54 Kb), 9 (1.42%; 2,010.62 Kb), 15 (1.23%; 1,264.4 Kb), 17
(1.19%; 970.16 Kb), 12 (1.17%; 1,559.9 Kb), and 6 (1.15%; 1,968.22 Kb)
[3]. Ten novel private pathogenic Copy Number Variations (CNVs) in 10
early-onset familial Alzheimer’s disease (EO-FAD) families overlapping
a set of genes (A2BP1, ABAT, CDH2, CRMP1, DMRT1, EPHA5, EPHA6,
ERMP1, EVC, EVC2, FLJ35024 and VLDLR) have also been identified [3].
Multiple polymorphic risk variants can increase neuronal vulnerability
to premature death. Among these susceptibility genes, the apolipoprotein
E (APOE) gene (19q13.2) (AD2) is the most prevalent as a risk factor for
AD, especially in those subjects harboring the APOE-4 allele [14], whereas
carriers of the APOE-2 allele are prone to longevity [15] and might be
protected against dementia [16-18].
APOE is the prototypical paradigm of a pleiotropic gene with
multifaceted activities in physiological and pathological conditions
[1,19]. ApoE is consistently associated with the amyloid plaque marker
for AD. APOE-4 may influence AD pathology by interacting with APP
metabolism and Aβ accumulation, enhancing hyperphosphorylation of
tau protein and Neurofibrillary Tangle (NFT) formation, reducing choline
acetyltransferase activity, increasing oxidative processes, modifying
inflammation-related neuroimmunotrophic activity and glial activation,
altering lipid metabolism, lipid transport and membrane biosynthesis
in sprouting and synaptic remodeling, and inducing neuronal apoptosis
[1,19-22]. In addition, multiple studies over the past two decades have
demonstrated that APOE variants may affect the therapeutic response to
anti-dementia drugs [21-30].
The distribution of APOE genotypes in the Iberian peninsula is as
follows: APOE-2/2 0.32%, APOE-2/3 7.3%, APOE-2/4 1.27%, APOE-3/3
71.11%, APOE-3/4 18.41%, and APOE-4/4 1.59% [7] (Figure 1). These
frequencies are very similar in Europe and in other Western societies.
There is a clear accumulation of APOE-4 carriers among patients with AD
(APOE-3/4 30.30%; APOE-4/4 6.06%) and VD (APOE-3/4 35.85%, APOE-
4/4 6.57%) as compared to controls (Figure 1).

Figure 1: Distribution and frequency of APOE genotypes in CNS
disorders.
(C: Controls, N=315; ANX: Anxiety, N=285; DEP: Depression, N=419;
PSY: Psychosis, N=162; STR: Stroke, N=67; AD: Alzheimer’s disease,
N=231; PAR: Parkinson’s disease, N=73; ADHD: Attention Deficit
Hyperactivity Disorder, N=42; MIG: Migraine, N=217; EPI: Epilepsy,
N=71; VD: Vascular dementia, N=198; VE: Vascular encephalopathy,
N=380; MS: Multiple sclerosis, N=21; CVI: Cerebrovascular insufficiency,
N=138; BT: Brain tumor, N=11; CNN: Cranial nerve neuropathy, N=25; MR:
Mental retardation, N=115; PTBS: Post-traumatic brain syndrome, N=59).
Significant differences (p<0.001) in the frequency of APOE genotypes
with respect to controls were only found in patients with Alzheimer’s
disease and vascular dementia. Significant differences were also found
between ANX vs. AD and VD (p<0.001), DEP vs. AD and VD (p<0.001),
PSY vs. AD (p<0.005), PSY vs. VD (p<0.001), PSY vs.VE (p<0.04), AD
vs. MIG (p<0.03), AD vs. VE (p<0.001), AD vs. PTBS (p<0.03), MIG vs.
VD (p<0.002), VD vs. VE (p<0.001), VD vs. PTBS (p<0.008), and VE
vs. MS (p<0.05)[205].
From studies designed to define APOE-related AD phenotypes, several
conclusions can be drawn: (i) the age-at-onset is 5-10 years earlier in
approximately 80% of AD cases harboring the APOE-4/4 genotype; (ii)
the serum levels of ApoE are lowest in APOE-4/4, intermediate in APOE-
3/3 and APOE-3/4, and highest in APOE-2/3 and APOE-2/4; (iii) serum
cholesterol levels are higher in APOE-4/4 than in the other genotypes; (iv)
HDL-cholesterol levels tend to be lower in APOE-3 homozygotes than
in APOE-4 allele carriers; (v) LDL-cholesterol levels are systematically
higher in APOE-4/4 than in any other genotype; (vi) triglyceride levels are
significantly lower in APOE-4/4; (vii) nitric oxide levels are slightly lower
in APOE-4/4; (viii) serum and cerebrospinal fluid (CSF) Aβ levels tend to
differ between APOE-4/4 and the other most frequent genotypes (APOE-
3/3, APOE-3/4); (ix) blood histamine levels are dramatically reduced
in APOE-4/4 as compared with the other genotypes; (x) brain atrophy
and AD neuropathology is markedly increased in APOE-4/4>APOE-
3/4>APOE-3/3; (xi) brain mapping activity shows a significant increase
in slow wave activity in APOE-4/4 from early stages of the disease; (xii)
brain hemodynamics, as reflected by reduced brain blood flow velocity
and increased pulsatility and resistance indices, is significantly worse in
APOE-4/4 (and in APOE-4 carriers in general, as compared with APOE-
3 carriers); brain hypoperfusion and neocortical oxygenation is also more
deficient in APOE-4 carriers; (xiii) lymphocyte apoptosis is markedly
enhanced in APOE-4 carriers; (xiv) cognitive deterioration is faster in
APOE-4/4 patients than in carriers of any other APOE genotype; (xv)
in approximately 3-8% of the AD cases, the presence of some dementiarelated
metabolic dysfunctions accumulates more in APOE-4 carriers than
in APOE-3 carriers; (xvi) some behavioral disturbances, alterations in
circadian rhythm patterns, and mood disorders are slightly more frequent
in APOE-4 carriers; (xvii) aortic and systemic atherosclerosis is also more
frequent in APOE-4 carriers; (xviii) liver metabolism and transaminase
activity also differ in APOE-4/4 with respect to other genotypes; (xix)
hypertension and other cardiovascular risk factors also accumulate
in APOE-4; and (xx) APOE-4/4 carriers are the poorest responders to
conventional drugs. These 20 major phenotypic features clearly illustrate
the biological disadvantage of APOE-4 homozygotes and the potential
consequences that these patients may experience when they receive
pharmacological treatment for AD and/or concomitant pathologies
[1,7,19-33].
In over 100 clinical trials for dementia, APOE has been used as the
only gene of reference for the pharmacogenomics of AD [1,7,21,22,26-
28,34-38]. Several studies indicate that the presence of the APOE-4
allele differentially affects the quality and extent of drug responsiveness
in AD patients treated with cholinergic enhancers (tacrine, donepezil,
galantamine, rivastigmine), neuroprotective compounds (nootropics),
endogenous nucleotides (CDP-choline), immunotrophins (anapsos),
neurotrophic factors (cerebrolysin), rosiglitazone or combination
therapies [39-41]; however, controversial results are frequently found
due to methodological problems, study design, and patient recruitment
in clinical trials. The major conclusion in most studies is that APOE-4
carriers are the worst responders to conventional treatments. When
APOE and CYP2D6 genotypes are integrated in bigenic clusters and the
APOE+CYP2D6-related therapeutic response to a combination therapy is
analyzed in AD patients, it becomes clear that the presence of the APOE-
4/4 genotype is able to convert pure CYP2D6*1/*1 extensive metabolizers
into full poor responders to conventional treatments, indicating the
existence of a powerful influence of the APOE-4 homozygous genotype on
the drug-metabolizing capacity of pure CYP2D6 extensive metabolizers.
In addition, a clear accumulation of APOE-4/4 genotypes is observed
among CYP2D6 poor and ultra-rapid metabolizers [26].
Different APP and PSEN1 and PSEN2 mutations may also modify the
therapeutic response to drugs acting on the amyloid cascade [42].
APOE-TOMM40 association
The TOMM40 locus is located adjacent to and in linkage disequilibrium
with APOE on 19q13.2. A poly T repeat in an intronic polymorphism
(rs10524523) (intron 6) in the TOMM40 gene, which encodes an outer
mitochondrial membrane translocase involved in the transport of Aβ and
other proteins into mitochondria, has been implicated in AD [43-56], and
APOE-TOMM40 genotypes have been shown to modify disease risk and
age at onset of symptoms [45,48-51,57], although the latter assumption
needs replication due to contradictory results [51,58-60]. Linnertz et al.
[44] defined 3 allele groups for rs10524523 (‘523’), based on the number of
‘T’-residues: ‘Short’ (S, T ≤ 19), ‘Long’ (L, 20 ≤ T ≤ 29) and ‘Very Long’ (VL,
T ≥ 30). Roses et al. [50-52] reported that longer lengths of rs10524523 are
associated with a higher risk for Late Onset Alzheimer’s Disease (LOAD);
for APOE-3/4 patients who developed LOAD after 60 years of age,
individuals with long poly T repeats (19-39 nucleotides) linked to APOE-
3 develop LOAD on an average of 7 years earlier than individuals with
shorter poly T repeats (11-16 nucleotides) linked to APOE-3 [45,49,50].
A fixed-effect meta-analysis approach showed that rs4420638 at the
TOMM40/APOE/APOC1 gene locus is associated with longevity [61,62].
Two independent associations with cognitive decline were found among
European-Americans in the 19q13.32 region (rs769449, APOE intron;
and rs115881343, TOMM40 intron); rs769449 was also associated with
cognitive decline among African-Americans, but rs115881343 was not
[63]. The APOE-TOMM40 genomic region is associated with cognitive
aging [64] and with pathological cognitive decline [65].
Linnertz et al. [66] investigated the genomic region spanning the
TOMM40 and APOE genes, to determine whether intronic poly T
(rs10524523) within this region affects expression of the APOE and
TOMM40 genes in the brain of patients with LOAD. The expression of
both genes was significantly increased with disease. Mean expression
of APOE and TOMM40 mRNA levels was higher in VL homozygotes
compared with S homozygotes in the temporal and occipital cortexes
from normal and LOAD cases. The 523 VL poly T resulted in significantly
higher expression than the S poly T. These results suggest that the
523 locus may contribute to LOAD susceptibility by modulating the
expression of TOMM40 and/or APOE transcription [66]. Recent studies
also suggest that the TOMM40 gene rs10524523 (“523”) variable length
poly T repeat polymorphism is associated to a certain extent with
similar AD phenotypes as those reported for APOE, such as brain white
matter changes [67,68] or different biomarkers [69-72]. In addition, the
TOMM40 rs2075650 G allele may be a risk factor for the development
of depression [73] and sporadic inclusion body myositis [74]. Different
markers at the 19q13-q13.2 chromosomal region, including the rs2075650
and rs157590 (TOMM40), rs1064725 (APOC1), and rs429358 and rs7412
(APOE) SNPs also show association with primary progressive aphasia and
the behavioral variant frontotemporal dementia [75].
The TOMM40/APOE/APOC1 loci have been associated with c-reactive
Protein (CRP), a heritable biomarker of systemic inflammation and
a predictor of Cardiovascular Disease (CVD) [76]. Genome-wide
Association Studies (GWAS) have identified LDL-cholesterol-associated
loci near HMGCR, ABO and TOMM40 [77], and also an association
of TOMM40 with blood lipid levels [78,79] and body mass index [80].
Genetic variants in TOMM40/APOE-C1-C2-C4 genes have also been
found to be associated with multiple cardiovascular-related traits [81-83].
We have investigated the structure of the APOE-TOMM40 region in
Spanish patients with dementia, and the influence of polymorphic variants
in this genomic segment on the therapeutic response to a multifactorial
treatment adapted to the pathogenic profile of the patients. The main aims
of the study were: (i) structural analysis of the APOE-TOMM40 region
(distribution and frequency of major genotypes, with special emphasis on
TOMM40 poly T variants) in the Spanish population with dementia; and
(ii) APOE- and TOMM40 poly T1/T2-related therapeutic response to a
multifactorial therapy in AD [84].
The distribution and frequency of APOE genotypes wereas follows:
APOE-2/3, 8.26%; APOE-2/4, 1.96%; APOE-3/3, 51.52%; APOE-3/4,
33.04%; and APOE-4/4, 5.22%. The distribution of 6 major TOMM40 poly
T variants was: 18.37% S/S, 7.83% S/L, 38.80% S/VL, 1.52% L/L, 7.17% L/
VL, and 26.31% VL/VL. The APOE-2/3 genotype was found to be associated
with S/S (27.63%), S/VL (51.32%), and L/VL (21.05%); APOE-2/4 was
associated with S/L (16.67%), S/VL (38.89%), L/VL (11.11%), and VL/VL
(33.33%); APOE-3/3 was associated with S/S (29.32%), S/L (0.42%), S/VL
(47.26%), L/VL (0.21%), and VL/VL (22.79%); APOE-3/4 was associated
with S/S (2.96%), S/L (21.38%), S/VL (28.29%), L/VL (15.46%), and VL/
VL (31.91%); and APOE-4/4 was associated with S/L (4.17%), S/VL (2.17%),
L/L (29.17%), L/VL (33.33%), and VL/VL (31.25%) (Figure 2). Likewise, the
S/S genotype was associated with APOE-2/3 (27.63%), 3/3 (29.32%), and
3/4 (2.96%); S/L with APOE-2/4 (16.67%), 3/3 (0.42%), 3/4 (21.38%), and
4/4 (4.17%); S/VL with APOE-2/3 (51.32%), 2/4 (38.89%), 3/3 (47.26%),
3/4 (28.29%), and 4/4 (2.08%); L/L was exclusively associated with APOE-
4/4 (100%); L/VL with APOE-2/4 (11.11%), 3/3 (0.21%), 3/4 (15.46%),
and 4/4 (33.33%); and VL/VL with APOE-2/3 (21.05%), 2/4 (33.33%), 3/3
(22.79%), 3/4 (31.91%), and 4/4 (31.25%) (Figure 3). S/VL and VL/VL
are the only TOMM40 poly T genotypes which interact with all major
APOE genotypes; in contrast, the APOE-4/4-TOMM40-L/L association is
unique, representing approximately 30% of APOE-4/4 carriers.The allele
distribution of TOMM40 poly T repeats in the Spanish population reflects a
high proportion of heterozygous S/VL (39%), followed by homozygous VL
(27%) and homozygous S (19%). Homozygous L/L represents 1.52% of the
Spanish population, and both S/VL and L/VL genotypes conform a group
of about 7-8% of the population. Potential dissimilarities with other White
and Hispanic populations [44] might be due to the ancestral admixture of
different cultures in the Iberian peninsula. The linkage pattern between
TOMM40-’523’ and APOE alleles in Whites and Hispanics reflects that
the L is primarily linked to APOE-4, while the majority of the VL and S are
linked to APOE-3. In African-Americans, Ghanaians and Japanese, there
is an increased frequency of the ‘523’S-APOE-4 [44].
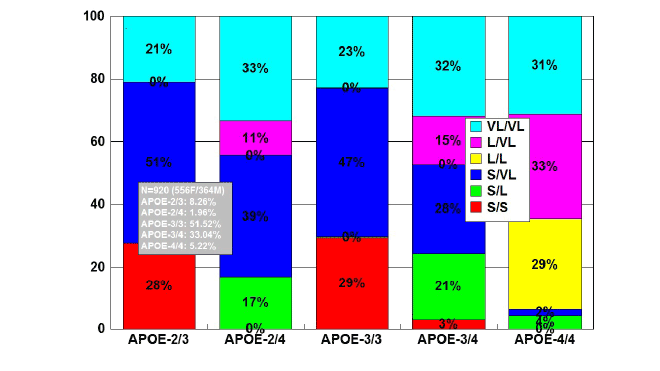
Figure 2: Distribution and frequency of TOMM40-Poly T variants
associated with APOE genotypes in patients with Alzheimer’s disease.
Patients with Alzheimer’s disease (N=920; 556 females, 364 males)
were classified according to their APOE genotype (APOE-2/3, 8.26%;
APOE-2/4, 1.96%; APOE-3/3, 51.52%; APOE-3/4, 33.04%; APOE-4/4,
5.22%) and the distribution and frequency of TOMM40-Poly T variants
(VL/VL, L/VL, L/L, S/VL, S/L, S/S) were studied in each APOE-related
group [84].
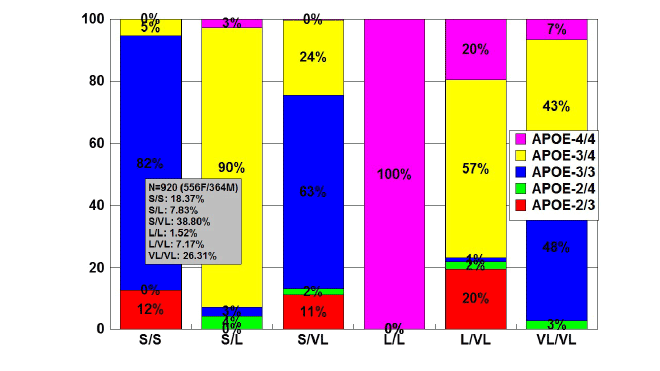
Figure 3: Distribution and frequency of APOE genotypes associated
with TOMM40-Poly T variants in patients with Alzheimer’s disease.
Patients with Alzheimer’s disease (N=920; 556 females, 364 males)
were classified according to their TOMM40-Poly T variants (S/S,
18.37%; S/L, 7.83%; S/VL, 38.80%; L/L, 1.52%; L/VL, 7.17%; VL/VL,
26.31%) and the distribution and frequency of APOE genotypes were
studied in each TOMM40-related group [84].
We found that patients harboring the APOE-4/4-L/L cluster developed
dementia at an earlier age (<70 yrs) than their counterparts with other
genotypes. In fact, L/L carriers were the youngest at age of onset, followed
by S/S carriers. In addition, virtually 100% of L/L carriers were exclusively
associated with APOE-4/4, representing the worst responders to our
combination therapy. The APOE-3/3-VL/VL cluster, with an earlier age
at onset (mean age ~70 yrs), was present in approximately a quarter of
APOE-3/3 carriers (Figure 2) [84].
In terms of therapeutic response to a combination therapy, a transient
profile of cognitive improvement for 6-12 months and maximum effect
during the first 3 months of treatment was observed in APOE-2/3, APOE-
2/4, APOE-3/3, APOE-3/4, and APOE-4/4 carriers, with significant effects
in APOE-3/3 carriers for 12 months. The response rate (RR) (MMSE score
after 12 months of treatment ≥ baseline MMSE score, prior to treatment)
was 70% in APOE-3/3, 67% in APOE-2/3, 56% in APOE-2/4, 50% in
APOE-4/4, and 45% in APOE-3/4 carriers, with significant differences
between APOE-2/3 and APOE-3/4, APOE-2/3 and APOE-4/4, APOE-3/3
and APOE-3/4, and APOE-3/3 and APOE-4/4.The time-dependent profile
of cognitive performance after treatment, according to the TOMM40
poly T genotype, was similar to that observed in the total group or in
the APOE-related study, with an apparent improvement during the first
3-9 months of treatment; however, significant effects were only observed
in patients harboring the TOMM40 poly T-S/S and S/VL genotypes. S/S
carriers were the best responders (70%), followed by S/VL (61%), VL/VL
(57%), and L/VL carriers (51%), and L/L (35%) and S/L carriers (45%)
were the worst responders [84].
Bernardi et al. [57] studied the association between TOMM40
rs10524523, age of onset, and memory performance in patients with the
PSEN1 M146L mutation in a large familial AD Calabrian kindred, and
found that APOE33/TOMM40VL/VL patients showed a tendency for an
earlier age at onset compared to those with APOE33/TOMM40VL/S and
APOE33/TOMM40S/S. TOMM40VL/VL patients had better memory
performance, when compared to TOMM40S/S but not to TOMM40VL/S
patients. For Li et al. [60], TOMM40 intron 6 poly T length may explain
some of the variation in age at onset in PSEN2 familial AD and may be
associated with AD neuropathology in persons with APOE-3/3.
Several reports suggest that both APOE and TOMM40 influence
memory performance in normal [64] and pathological conditions [65,85].
For some authors, both TOMM40 and APOE significantly influence agerelated
memory performance, but they appear to do so independently
of each other [85]. Others suggest important APOE-independent
associations between the TOMM40 ‘523’ polymorphism and specific
cognitive domains of memory and executive control that are preferentially
affected in early-stage AD, with S homozygotes performing better than
the S/L-S/VL and the VL/L-L/VL-VL/VL genotype groups on measures
associated with memory and executive function [65]. According to our
data, the best mental performance (and response rate to treatment) is
observed in patients harboring the APOE-3/3-S/S haplotype (R~70%),
followed by those with the APOE-3/3-S/VL haplotype (R~60%). In
general, S/S carriers are the best responders > S/VL (61%) > VL/VL
(57%) > L/VL (51%) > S/L (45%) > L/L (35%). The presence of the L allele
appears to contribute to a poor therapeutic outcome, and when the L/L
genotype associates with the APOE-4/4 genotype, carriers of the APOE-
4/4-S/S haplotype (30% of APOE-4/4 carriers) are converted into the
worst responders) [84].
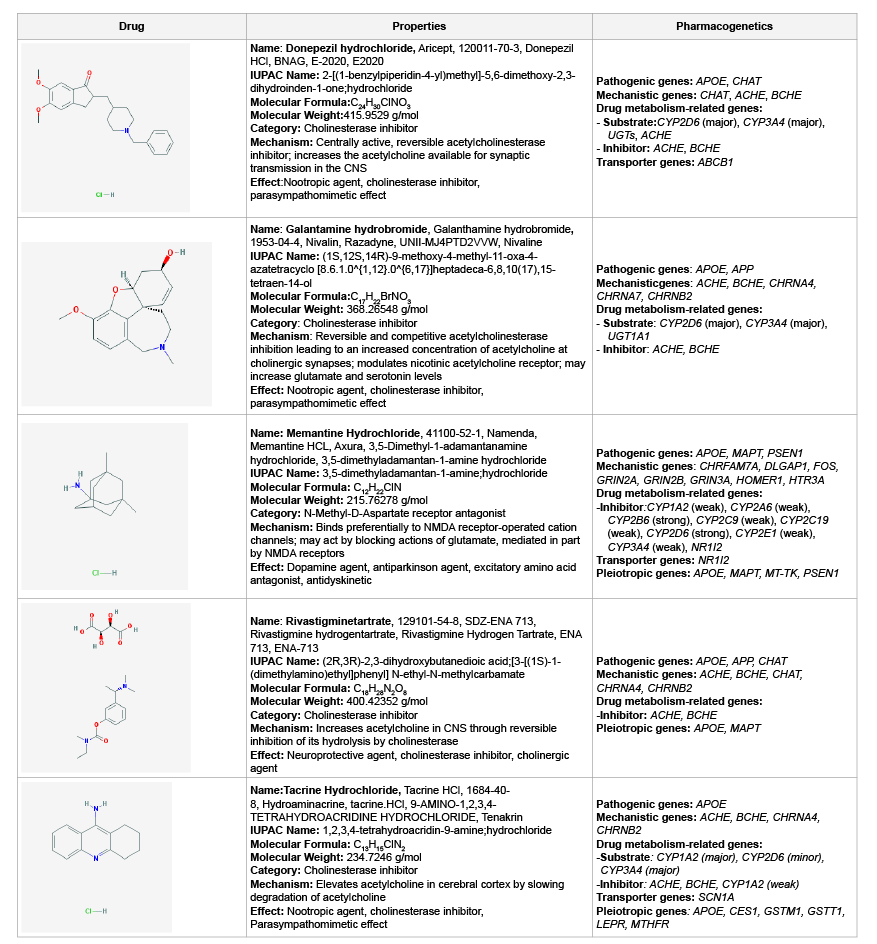
Genes involved in the mechanism of action of CNS drugs
Most genes associated with the mechanism of action of Central Nervous
System (CNS) drugs encode receptors, enzymes, and neurotransmitters on
which psychotropic drugs act as ligands (agonists, antagonists), enzyme
modulators (substrates, inhibitors, inducers) or neurotransmitter regulators
(releasers, reuptake inhibitors) [8]. In the case of conventional anti-dementia
drugs, tacrine, donepezil, rivastigmine and galantamine are cholinesterase
inhibitors; and memantine is a partial N-Methyl-D-Aspartat (NMDA)
antagonist [3,86] (Table 1).
Genes involved in drug metabolism
Drug metabolism includes phase I reactions (i.e. oxidation, reduction,
hydrolysis) and phase II conjugation reactions (i.e. acetylation,
glucuronidation, sulphation, methylation). The principal enzymes with
polymorphic variants involved in phase I reactions are the following:
Cytochrome P450 monooxygenases (CYP3A4/5/7, CYP2E1, CYP2D6,
CYP2C19, CYP2C9, CYP2C8, CYP2B6, CYP2A6, CYP1B1, CYP1A1/2),
epoxide hydrolase, esterases, NQO1 (NADPH-quinone oxidoreductase),
DPD (dihydropyrimidine dehydrogenase), ADH (alcohol dehydrogenase),
and ALDH (aldehyde dehydrogenase); and major enzymes involved in
phase II reactions include UGTs (uridine 5’-triphosphate glucuronosyl
transferases), TPMT (thiopurine methyltransferase), COMT (catecholO-methyltransferase),
HMT (histamine methyl-transferase), STs
(sulfotransferases), GST-A (glutathione S-transferase A), GST-P, GST-T,
GST-M, NAT1 (N-acetyl transferase 1), NAT2, and others. Among these
enzymes, CYP2D6, CYP2C9, CYP2C19, and CYP3A4/5 are the most
relevant in the pharmacogenetics of CNS drugs [8,27]. Approximately,
18% of neuroleptics are major substrates of CYP1A2 enzymes, 40%
of CYP2D6, and 23% of CYP3A4; 24% of antidepressants are major
substrates of CYP1A2 enzymes, 5% of CYP2B6, 38% of CYP2C19, 85%
of CYP2D6, and 38% of CYP3A4; 7% of benzodiazepines are major
substrates of CYP2C19 enzymes, 20% of CYP2D6, and 95% of CYP3A4
[8,27]. Most CYP enzymes exhibit ontogenic-, age-, sex-, circadian-, and
ethnic-related differences [8,86].
ADH1A: Alcohol dehydrogenase 1A (class I), alpha polypeptide; AADAC: Arylacetamide deacetylase; AANAT: aralkylamine N-acetyltransferase; ACSL1: AcylCoA
synthetase long-chain family member 1; ACSL3: Acyl-CoA synthetase long-chain family member 3; ACSL4: Acyl-CoA synthetase long-chain family member 4;
ACSM1: Acyl-CoA synthetase medium-chain family member 1; ACSM2B: Acyl-CoA synthetase medium-chain family member 2B; ACSM3: Acyl-CoA synthetase
medium-chain family, member 3; ADH1B: Alcohol dehydrogenase 1B (class I), beta polypeptide; ADH1C: Alcohol dehydrogenase 1C (class I), gamma polypeptide;
ADH4: Alcohol dehydrogenase 4 (class II), pi polypeptide; ADH5: Alcohol dehydrogenase 5 (class III), chi polypeptide; ADH6: Alcohol dehydrogenase 6 (class V);
ADH7: Alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide; ADHFE1: Alcohol dehydrogenase, iron containing, 1; AGXT: Alanine-glyoxylate aminotransferase;
AKR1A1: Aldo-keto reductase family 1, member A1 (aldehyde reductase); AKR1B1: Aldo-keto reductase family 1, member B1 (aldose reductase); AKR1C1: Aldoketo
reductase family 1, member C1; AKR1D1: Aldo-keto reductase family 1, member D1; ALDH1A1: Aldehyde dehydrogenase 1 family, member A1; ALDH1A2:
Aldehyde dehydrogenase family 1, subfamily A2; ALDH1A3: Aldehyde dehydrogenase family 1, subfamily A3; ALDH1B1: Aldehyde dehydrogenase 1 family, member
B1; ALDH2: Aldehyde dehydrogenase 2 family (mitochondrial); ALDH3A1: Aldehyde dehydrogenase 3 family, member A1; ALDH3A2: Aldehyde dehydrogenase 3
family, member A2; ALDH3B1: Aldehyde dehydrogenase 3 family, member B1; ALDH3B2: Aldehyde dehydrogenase 3 family, member B2; ALDH4A1: Aldehyde
dehydrogenase 4 family, member A1; ALDH5A1: Aldehyde dehydrogenase 5 family, member A1; ALDH6A1: Aldehyde dehydrogenase 6 family, member A1;
ALDH7A1: Aldehyde dehydrogenase 7 family, member A1; ALDH8A1: Aldehyde dehydrogenase 8 family, member A1; ALDH9A1: Aldehyde dehydrogenase 9
family, member A1; AOX1: Aldehyde oxidase 1; AS3MT: Arsenic (+3 oxidation state) methyltransferase; ASMT: Acetylserotonin O-methyltransferase; BAAT: Bile
acid CoA: amino acid N-acyltransferase (glycine N-choloyltransferase); CBR1: Carbonyl reductase 1; CBR3: Carbonyl reductase 3; CBR4: Carbonyl reductase 4;
CCBL1: Cysteine conjugate-beta lyase, cytoplasmic; CDA: Cytidine deaminase; CEL: Carboxyl ester lipase; CES1: Carboxylesterase 1; CES1P1: Carboxylesterase
1 pseudogene 1; CES2: Carboxylesterase 2; CES3: Carboxylesterase 3; CES5A: Carboxylesterase 5A; CHST1: Carbohydrate (keratan sulfate Gal-6) sulfotransferase
1; CHST2: Carbohydrate (N-acetylglucosamine-6-O) sulfotransferase 2; CHST3: Carbohydrate (chondroitin 6) sulfotransferase 3; CHST4: Carbohydrate
(N-acetylglucosamine 6-O) sulfotransferase 4; CHST5: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 5; CHST6: Carbohydrate (N-acetylglucosamine
6-O) sulfotransferase 6; CHST7: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 7; CHST8: Carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase
8; CHST9: Carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 9; CHST10: Carbohydrate sulfotransferase 10; CHST11: Carbohydrate (chondroitin 4)
sulfotransferase 11; CHST12: Carbohydrate (chondroitin 4) sulfotransferase 12; CHST13: Carbohydrate (chondroitin 4) sulfotransferase 13; COMT: Catechol-Omethyltransferase;
CYB5R3: Cytochrome b5 reductase 3; CYP1A1: Cytochrome P450, family 1, subfamily A, polypeptide 1; CYP1A2: Cytochrome P450, family 1,
subfamily A, polypeptide 2; CYP1B1: Cytochrome P450, family 1, subfamily B, polypeptide 1; CYP2A6: Cytochrome P450, family 2, subfamily A, polypeptide 6;
CYP2A7: Cytochrome P450, family 2, subfamily A, polypeptide 7; CYP2A13: Cytochrome P450, family 2, subfamily A, polypeptide 13; CYP2B6: Cytochrome P450,
family 2, subfamily B, polypeptide 6; CYP2C8: Cytochrome P450, family 2, subfamily C, polypeptide 8; CYP2C9: Cytochrome P450, family 2, subfamily C, polypeptide
9; CYP2C18: Cytochrome P450, family 2, subfamily C, polypeptide 18; CYP2C19: Cytochrome P450, family 2, subfamily C, polypeptide 19; CYP2D6: Cytochrome
P450, family 2, subfamily D, polypeptide 6; CYP2D7P1: Cytochrome P450, family 2, subfamily D, polypeptide 7 pseudogene 1; CYP2E1: Cytochrome P450, family
2, subfamily E, polypeptide 1; CYP2F1: Cytochrome P450, family 2, subfamily F, polypeptide 1; CYP2J2: Cytochrome P450, family 2, subfamily J, polypeptide 2;
CYP2R1: Cytochrome P450, family 2, subfamily R, polypeptide 1; CYP2S1: Cytochrome P450, family 2, subfamily S, polypeptide 1; CYP2W1: Cytochrome P450,
family 2, subfamily W, polypeptide 1; CYP3A4: Cytochrome P450, family 3, subfamily A, polypeptide 4; CYP3A5: Cytochrome P450, family 3, subfamily A, polypeptide
5; CYP3A7: Cytochrome P450, family 3, subfamily A, polypeptide 7; CYP3A43: Cytochrome P450, family 3, subfamily A, polypeptide 43; CYP4A11: Cytochrome
P450, family 4, subfamily A, polypeptide 11; CYP4A22: Cytochrome P450, family 4, subfamily A, polypeptide 22; CYP4B1: Cytochrome P450, family 4, subfamily B,
polypeptide 1; CYP4F2: Cytochrome P450, family 4, subfamily F, polypeptide 2; CYP4F3: Cytochrome P450, family 4, subfamily F, polypeptide 3; CYP4F8:
Cytochrome P450, family 4, subfamily F, polypeptide 8; CYP4F11: Cytochrome P450, family 4, subfamily F, polypeptide 11; CYP4F12: Cytochrome P450, family 4,
subfamily F, polypeptide 12; CYP4Z1: Cytochrome P450, family 4, subfamily Z, polypeptide 1; CYP7A1: Cytochrome P450, family 7, subfamily A, polypeptide 1;
CYP7B1: Cytochrome P450, family 7, subfamily B, polypeptide 1; CYP8B1: Cytochrome P450, family 8, subfamily B, polypeptide 1; CYP11A1: Cytochrome P450,
family 11, subfamily A, polypeptide 1; CYP11B1: Cytochrome P450, family 11, subfamily B, polypeptide 1: CYP11B2: Cytochrome P450, family 11, subfamily B,
polypeptide 2; CYP17A1: Cytochrome P450, family 17, subfamily A, polypeptide 1; CYP19A1: Cytochrome P450, family 19, subfamily A, polypeptide 1; CYP20A1:
Cytochrome P450, family 20, subfamily A, polypeptide 1; CYP21A2: Cytochrome P450, family 21, subfamily A, polypeptide 2; CYP24A1: Cytochrome P450, family
24, subfamily A, polypeptide 1; CYP26A1: Cytochrome P450, family 26, subfamily A, polypeptide 1; CYP26B1: Cytochrome P450, family 26, subfamily B, polypeptide
1; CYP26C1: Cytochrome P450, family 26, subfamily C, polypeptide 1; CYP27A1: Cytochrome P450, family 27, subfamily A, polypeptide 1; CYP27B1: Cytochrome
P450, family 27, subfamily B, polypeptide 1; CYP39A1: Cytochrome P450, family 39, subfamily A, polypeptide 1; CYP46A1: Cytochrome P450, family 46, subfamily
A, polypeptide 1; CYP51A1: Cytochrome P450, family 51, subfamily A, polypeptide 1; DDOST: Dolichyl-diphosphooligosaccharide--protein glycosyltransferase
subunit (non-catalytic); DHRS1: Dehydrogenase/reductase (SDR family) member 1; DHRS2: Dehydrogenase/reductase (SDR family) member 2; DHRS3:
Dehydrogenase/reductase (SDR family) member 3; DHRS4: Dehydrogenase/reductase (SDR family) member 4; DHRS7: Dehydrogenase/reductase (SDR family)
member 7; DHRS9: Dehydrogenase/reductase (SDR family) member 9; DHRS12: Dehydrogenase/reductase (SDR family) member 12; DHRS13: Dehydrogenase/
reductase (SDR family) member 13; DHRSX: Dehydrogenase/reductase (SDR family) X-linked; DLGAP1: discs, large (Drosophila) homolog-associated protein 1;
DPEP1: Dipeptidase 1 (renal); DPYD: Dihydropyrimidine dehydrogenase; EPHX1: Epoxide hydrolase 1, microsomal (xenobiotic); EPHX2: Epoxide hydrolase 2,
microsomal (xenobiotic); ESD: Esterase D; FMO1: Flavin containing monooxygenase 1; FMO2: Flavin containing monooxygenase 2; FMO3: Flavin containing
monooxygenase 3; FMO4: Flavin containing monooxygenase 4; FMO5: Flavin containing monooxygenase 5; FMO6P: Flavin containing monooxygenase 6
pseudogene; FOS: FBJ murine osteosarcoma viral oncogene homolog; GAL3ST1: Galactose-3-O-sulfotransferase 1; GAMT: Guanidinoacetate N-methyltransferase;
GLRX: Glutaredoxin (thioltransferase); GLYAT: Glycine-N-acyltransferase; GNMT: Glycine N-methyltransferase; GPX1: Glutathione peroxidase 1; GPX2: Glutathione
peroxidase 2 (gastrointestinal); GPX3: Glutathione peroxidase 3 (plasma); GPX4: Glutathione peroxidase 4; GPX5: Glutathione peroxidase 5; GPX6: Glutathione
peroxidase 6 (olfactory); GPX7: Glutathione peroxidase 7; GSR: Glutathione reductase; GSTA1: Glutathione S-transferase alpha 1; GSTA2: Glutathione S-transferase
alpha 2; GSTA3: Glutathione S-transferase alpha 3; GSTA4: Glutathione S-transferase alpha 4; GSTA5: Glutathione S-transferase alpha 5; GSTCD: Glutathione
S-transferase, C-terminal domain containing; GSTK1: Glutathione S-transferase kappa 1; GSTM1: Glutathione S-transferase mu 1; GSTM2: Glutathione S-transferase
mu 2 (muscle); GSTM3: Glutathione S-transferase mu 3 (brain); GSTM4: Glutathione S-transferase mu 4; GSTM5: Glutathione S-transferase mu 5; GSTO1:
Glutathione S-transferase omega 1; GSTO2: Glutathione S-transferase omega 2; GSTP1: Glutathione S-transferase pi 1; GSTT1: Glutathione S-transferase theta
1; GSTT2: Glutathione S-transferase theta 2; GSTZ1: Glutathione S-transferase zeta 1; GZMA: Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine
esterase 3; GZMB: Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1); HNMT: Histamine N-methyltransferase; HOMER1: homer
homolog 1 (Drosophila); HSD11B1: Hydroxysteroid (11-beta) dehydrogenase 1; HSD17B10: Hydroxysteroid (17-beta) dehydrogenase 10; HSD17B11: Hydroxysteroid
(17-beta) dehydrogenase 11; HSD17B14: Hydroxysteroid (17-beta) dehydrogenase 14; INMT: Indolethylamine N-methyltransferase; MAOA: Monoamine oxidase A;
MAOB: monoamine oxidase B; METAP1: Methionyl aminopeptidase 1; MGST1: Microsomal glutathione S-transferase 1; MGST2: Microsomal glutathione
S-transferase 1; MGST3: Microsomal glutathione S-transferase 3; NAA20: N(alpha)-acetyltransferase 20, NatB catalytic subunit; NAT1: N-acetyltransferase 1
(arylamine N-acetyltransferase); NAT2: N-acetyltransferase 2 (arylamine N-acetyltransferase); NNMT: Nicotinamide N-methyltransferase; NQO1: NAD(P)H
dehydrogenase, quinone 1; NQO2: NAD(P)H dehydrogenase, quinone 2; NR1I2:nuclear receptor subfamily 1, group I, member 2; PNMT: Phenylethanolamine
N-methyltransferase; PON1: Paraoxonase 1; PON2: Paraoxonase 2; PON3: Paraoxonase 3; POR: P450 (cytochrome) oxidoreductase; PTGES: Prostaglandin E
synthase; PTGS1: Prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase); PTGS2: Prostaglandin-endoperoxide synthase 2
(prostaglandin G/H synthase and cyclooxygenase); SAT1: Spermidine/spermine N1-acetyltransferase 1; SMOX: Spermine oxidase; SOD1: Superoxide dismutase
1, soluble; SOD2: Superoxide dismutase 2, mitochondrial; SULT1A1: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 1; SULT1A2: Sulfotransferase
family, cytosolic, 1A, phenol-preferring, member 2; SULT1A3: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 3; SULT1B1: Sulfotransferase family,
cytosolic, 1B, member 1; SULT1C1: Sulfotransferase family, cytosolic, 1C, member 1; SULT1C2: Sulfotransferase family, cytosolic, 1C, member 2; SULT1C3:
Sulfotransferase family, cytosolic, 1C, member 3; SULT1C4: Sulfotransferase family, cytosolic, 1C, member 4; SULT1E1: Sulfotransferase family 1E, estrogenpreferring,
member 1; SULT2A1: Sulfotransferase family, cytosolic, 2A, dehydroepiandrosterone (DHEA)-preferring, member 1; SULT2B1: Sulfotransferase family,
cytosolic, 2B, member 1; SULT4A1: Sulfotransferase family 4A, member 1; SULT6B1: sulfotransferase family, cytosolic, 6B, member 1; TBXAS1: Thromboxane A
synthase 1 (platelet); TPMT: Thiopurine S-methyltransferase; TST: Thiopurine S-methyltransferase; UCHL1: Ubiquitin carboxyl-terminal esterase L1 (ubiquitin
thiolesterase); UCHL3: Ubiquitin carboxyl-terminal esterase L3 (ubiquitin thiolesterase); UGT1A1: UDP glucuronosyltransferase 1 family, polypeptide A1; UGT1A3:
UDP glucuronosyltransferase 1 family, polypeptide A3; UGT1A4: UDP glucuronosyltransferase 1 family, polypeptide A4; UGT1A5: UDP glucuronosyltransferase 1
family, polypeptide A5; UGT1A6: UDP glucuronosyltransferase 1 family, polypeptide A6; UGT1A7: UDP glucuronosyltransferase 1 family, polypeptide A7; UGT1A8:
UDP glucuronosyltransferase 1 family, polypeptide A8; UGT1A9: UDP glucuronosyltransferase 1 family, polypeptide A9; UGT1A10: UDP glucuronosyltransferase 1
family, polypeptide A10; UGT2A1: UDP glucuronosyltransferase 2 family, polypeptide A1, complex locus; UGT2A3: UDP glucuronosyltransferase 2 family, polypeptide
A3; UGT2B10: UDP glucuronosyltransferase 2 family, polypeptide B10; UGT2B11: UDP glucuronosyltransferase 2 family, polypeptide B11; UGT2B15: UDP
glucuronosyltransferase 2 family, polypeptide B15; UGT2B17: UDP glucuronosyltransferase 2 family, polypeptide B17; UGT2B28: UDP glucuronosyltransferase 2
family, polypeptide B28; UGT2B4: UDP glucuronosyltransferase 2 family, polypeptide B4; UGT2B7: UDP glucuronosyltransferase 2 family, polypeptide B7; UGT3A1:
UDP glycosyltransferase 3 family, polypeptide A1; UGT8: UDP glycosyltransferase 8; XDH: Xanthine dehydrogenase.
Source [Ref.86]
Table 1: Pharmacogenomics of conventional anti-dementia drugs
In dementia, as in any other CNS disorders, CYP genomics is a highly
important issue, since in practice over 90% of patients with dementia are
daily consumers of psychotropics. Furthermore, some acetylcholinesterase
inhibitors (the most prescribed anti-dementia drugs worldwide) are
metabolized via CYP enzymes (Table 1). Most CYP enzymes display highly
significant ethnic differences, indicating that the enzymatic capacity of
these proteins varies depending upon the polymorphic variants present in
their coding CYP genes. The practical consequence of this genetic variation
is that the same drug can be differentially metabolized according to the
genetic profile of each subject, and that knowing the pharmacogenomic
profile of an individual, his/her pharmacodynamic response is potentially
predictable. This is the cornerstone of pharmacogenetics. In this regard,
the CYP2D6, CYP2C19, CYP2C9 and CYP3A4/5 genes and their respective
protein products deserve special consideration.
CYP2D6
CYP2D6 is a 4.38 kb gene with 9 exons mapped on 22q13.2. Four RNA
transcripts of 1190-1684 bp are expressed in the brain, liver, spleen and
reproductive system where 4 major proteins of 48-55 kDa (439-494aa)
are identified. This protein is a transport enzyme of the cytochrome
P450 subfamily IID or multigenic cytochrome P450 superfamily of
mixed-function monooxygenases. The cytochrome P450 proteins are
monooxygenases which catalyze many reactions involved in drug
metabolism and synthesis of cholesterol, steroids and other lipids.
This protein localizes to the endoplasmic reticulum and is known to
metabolize as many as 25% of commonly prescribed drugs and over
60% of current psychotropics. Its substrates include debrisoquine,
an adrenergic-blocking drug; sparteine and propafenone, both antiarrhythmic
drugs; and amitryptiline, an anti-depressant. The gene is
highly polymorphic in the population. There are 141 CYP2D6 allelic
variants of which -100C>T, -1023C>T, -1659G>A, -1707delT, -1846G>A,
-2549delA, -2613-2615delAGA, -2850C>T, -2988G>A, and -3183G>A
represent the 10 most important variants [86-88]. Different alleles
result in the extensive, intermediate, poor, and ultra-rapid metabolizer
phenotypes, characterized by normal, intermediate, decreased, and
multiplied ability to metabolize the enzyme’s substrates, respectively. The
hepatic cytochrome P450 system is responsible for the first phase in the
metabolism and elimination of numerous endogenous and exogenous
molecules and ingested chemicals. P450 enzymes convert these substances
into electrophilic intermediates which are then conjugated by phase II
enzymes (e.g. UDP glucuronosyltransferases, N-acetyltransferases) to
hydrophilic derivatives that can be excreted. According to the database of
the World Guide for Drug Use and Pharmacogenomics variants [86], 982
drugs are CYP2D6-related: 371 drugs are substrates, over 300 drugs are
inhibitors, and 18 drugs are CYP2D6 inducers.
In healthy subjects, Extensive Metabolizers (EMs) account for 55.71%
of the population, whereas Intermediate Metabolizers (IMs) account for
34.7%, Poor Metabolizers (PMs) 2.28%, and Ultra-rapid Metabolizers
(UMs) 7.31%. Remarkable interethnic differences exist in the frequency
of the PM and UM phenotypes among different societies all over the
world [89-91]. On average, approximately 6.28% of the world population
belongs to the PM category. Europeans (7.86%), Polynesians (7.27%),
and Africans (6.73%) exhibit the highest rate of PMs, whereas Orientals
(0.94%) show the lowest rate [89]. The frequency of PMs among Middle
Eastern populations, Asians, and Americans is in the range of 2-3%.
CYP2D6 gene duplications are relatively infrequent among Northern
Europeans, but in East Africa the frequency of alleles with duplication of
CYP2D6 is as high as 29% [92]. In Europe, there is a North-South gradient
in the frequency of PMs (6-12% of PMs in Southern European countries,
and 2-3% PMs in Northern latitudes) [8].
In AD, EMs, IMs, PMs, and UMs are 56.38%, 27.66%, 7.45%, and 8.51%,
respectively, and in VD, 52.81%, 34.83%, 6.74%, and 5.62%, respectively
(Figure 4 and Figure 5). There is an accumulation of AD-related genes
of risk in PMs and UMs. EMs and IMs are the best responders, and
PMs and UMs are the worst responders to a combination therapy with
cholinesterase inhibitors, neuroprotectants, and vasoactive substances.
The pharmacogenetic response in AD appears to be dependent upon
the networking activity of genes involved in drug metabolism and genes
involved in AD pathogenesis [1,7,25-28,38,93].
APOE-CYP2D6 association
In a population of 582 Spanish patients with AD (APOE-2/3 0.34%;
APOE-2/3 9.28%; APOE-2/4 1.20%; APOE-3/359.45%; APOE-3/4 26.12%;
APOE-4/4 3.61%), CYP2D6-EMs represented 60.48%, IM 28.19%, PMs
5.32%, and UMs 6.01%. In APOE-2/2 carriers (N=2), 50% were EMs
and 50% IMs. In APOE-2/3 (N=54), 48.15% were EMs, 37.04% IMs,
1.85% PMs, and 12.96% UMs. In APOE-2/4 cases (N=7), 85.71% were
EMs and 14.29% IMs. In APOE-3/3 (N=346), 62.14% were EMs, 28.32%
IMs, 4.63% PMs, and 4.91% UMs. In APOE-3/4 (N=152), 61.18% were
EMs, 26.66% IMs, 7.90% PMs, and 5.26% UMs. In APOE-4/4 (N=21),
52.38% were EMs, 28.57% IMs, 4.76% PMs, and 14.29% UMs (Figure 6).
Significant differences were found in the distribution of CYP2D6 variants
between APOE-3/3 and APOE-2/3 carriers (p<0.01), and to a lesser
extent between APOE-3/3 and APOE-4/4 carriers (p: 0.06). A tendency
toward the accumulation of PMs and UMs in APOE-4 carriers was also
observed (Figure 7). The presence of CYP2D6 PMs and UMs in APOE-4
carriers may account for the poor response to conventional treatments
currently observed in those patients harboring the APOE-3/4 and APOE-
4/4 genotypes [3].
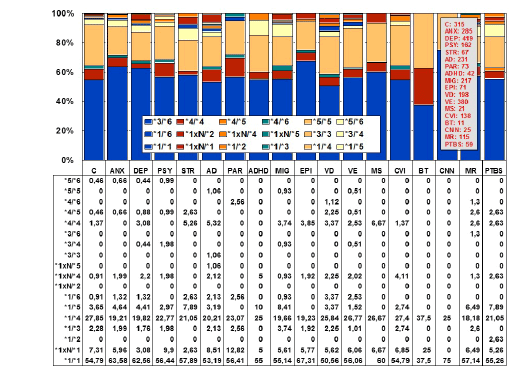
Figure 4: Distribution and frequency of CYP2D6 genotypes in CNS
disorders.
(C: Controls, N=315; ANX: Anxiety, N=285; DEP: Depression, N=419;
PSY: Psychosis, N=162; STR: Stroke, N=67; AD: Alzheimer’s disease,
N=231; PAR: Parkinson’s disease, N=73; ADHD: Attention Deficit
Hyperactivity Disorder, N=42; MIG: Migraine, N=217; EPI: Epilepsy,
N=71; VD: Vascular dementia, N=198; VE: Vascular encephalopathy,
N=380; MS: Multiple sclerosis, N=21; CVI: Cerebrovascular insufficiency,
N=138; BT: Brain tumor, N=11; CNN: Cranial nerve neuropathy, N=25;
MR: Mental retardation, N=115; PTBS: Post-traumatic brain syndrome,
N=59)[205].
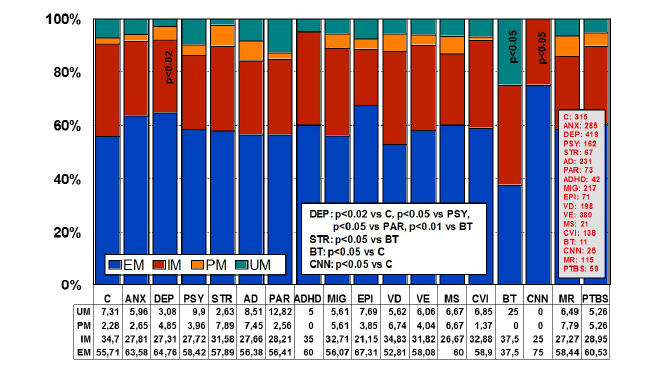
Figure 5: Distribution and frequency of CYP2D6 Extensive Metabolizers
(EM), Intermediate Metabolizers (IM), Poor Metabolizers (PM) and
Ultra-rapid Metabolizers (UM) in CNS disorders.
(C: Controls, N=315; ANX: Anxiety, N=285; DEP: Depression, N=419;
PSY: Psychosis, N=162; STR: Stroke, N=67; AD: Alzheimer’s disease,
N=231; PAR: Parkinson’s disease, N=73; ADHD: Attention Deficit
Hyperactivity Disorder, N=42; MIG: Migraine, N=217; EPI: Epilepsy,
N=71; VD: Vascular dementia, N=198; VE: Vascular encephalopathy,
N=380; MS: Multiple sclerosis, N=21; CVI: Cerebrovascular insufficiency,
N=138; BT: Brain tumor, N=11; CNN: Cranial nerve neuropathy, N=25;
MR: Mental retardation, N=115; PTBS: Post-traumatic brain syndrome,
N=59).
Significant differences were found between controls and DEP (p<0.02),
BT (p<0.05), and CNN (p<0.05). Patients with DEP also showed
differences with PSY (p<0.05), PAR (p<0.05), and BT (p<0.01); and
patients with STR exhibited significant differences with regard to BT
(p<0.05) [205].
CYP2C9
CYP2C9 is a gene (50.71 kb) with 9 exons mapped on 10q24. An RNA
transcript of 1860 bp is mainly expressed in hepatocytes where a protein
of 55.63 kDa (490 aa) can be identified. Over 600 drugs are CYP2C9-
related, 311 acting as substrates (177 are major substrates, 134 are minor
substrates), 375 as inhibitors (92 weak, 181 moderate, and 102 strong
inhibitors), and 41 as inducers of the CYP2C9 enzyme [86]. There are 481
CYP2C9 SNPs. By phenotypes, in the control population, PMs represent
7.04%, IMs 32.39%, and EMs 60.56%. In AD, PMs, IMs, and EMs are
6.45%, 37.64%, and 55.91% respectively, and in VD are 3.61%, 28.92%,
and 67.47% respectively [7] (Figure 8).
APOE-CYP2C9 association
In a sample of 566 Spanish patients with AD (APOE-2/2 0.35%; APOE-
2/39.01%; APOE-2/4 1.24%; APOE-3/3 59.10%; APOE-3/4 26.68%;
APOE-4/4 3.53%), 59.54% were CYP2C9-EMs, 35.34% CYP2C9-IMs, and
5.12% CYP2C9-PMs. By APOE genotype, 100% of homozygous APOE-2
(N=2) were EMs; in APOE-2/3 carriers (N=51), 43.14% were EMs, 49.02%
IMs, and 7.84% PMs. In APOE-2/4 carriers (N=7), 28.57% were EMs and
71.43% IMs; there were no PMs in the sample. In APOE-3/3 carriers
(N=335), 64.48% were EMs, 31.34% IMs, and 4.18% PMs. In APOE-3/4
carriers (N=151), 57.62% were EMs, 35.76% IMs, and 6.62% PMs; and
in APOE-4/4 carriers (N=20), 40% were EMs, 55% IMs, and 5% PMs
(Figure 9). There is an apparent reduction in the number of EMs among
APOE-2/3, APOE-3/4, and APOE-4/4 carriers, as compared with APOE-
3/3 carriers, and a correlative increase of IMs. The number of CYP2C9-
PMs is similar, ranging from 4.18% in APOE-3/3 carriers to 5% in APOE-
4/4, 6.62% in APOE-3/4, and 7.84% in APOE-2/3 carriers. There is a clear
accumulation of APOE-3/3 genotypes in CYP2C9-EMs, and of APOE-3/4
genotypes in CYP2C9-PMs, suggesting that the latter association might
also contribute to a poor pharmacogenetic outcome in AD patients, as
previously reported [3] (Figure10).
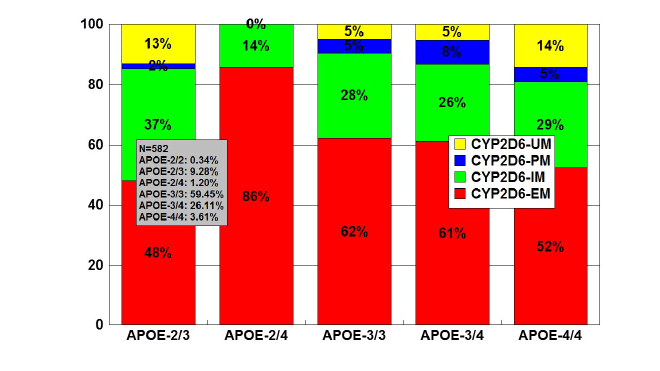
Figure 6: Distribution and frequency of CYP2D6 Extensive Metabolizers
(EM), Intermediate Metabolizers (IM), Poor Metabolizers (PM) and
Ultra-Rapid Metabolizers (UM) associated with APOE genotypes in
patients with Alzheimer’s disease.
Patients (N=582) were classified according to their APOE genotype
(APOE-2/2, 0.34%; APOE-2/3, 9.28%; APOE-2/4, 1.20%; APOE-3/3,
59.45%; APOE-3/4, 26.12%; APOE-4/4, 3.61%) and the distribution
and frequency of CYP2D6-EMs, IMs, PMs and UMs were studied in
each APOE-related group. Significant differences were found between
APOE-3/3 and APOE-2/3 (p<0.003), and a different pattern of CYP2D6
variants was also observed between APOE-3/3 and APOE-4/4 carriers
(p<0.06).
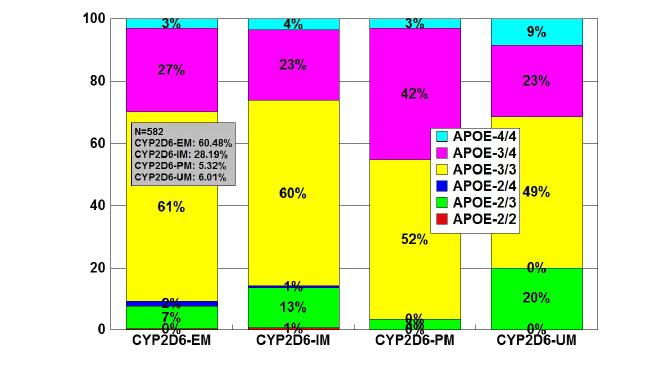
Figure 7: Distribution and frequency of APOE genotypes associated
with CYP2D6 Extensive Metabolizers (EM), Intermediate Metabolizers
(IM), Poor Metabolizers (PM) and Ultra-Rapid Metabolizers (UM) in
patients with Alzheimer’s disease.
Patients (N=582) were classified according to their CYP2D6 profile
(EMs: 60.48%, IMs: 28.19%; PMs: 5.32%; UMs: 6.01%), and the
distribution and frequency of APOE genotypes were studied in each
CYP2D6-related geno-phenotype.
Figure 8: Distribution and frequency of CYP2C9 Extensive Metabolizers
(EM), Intermediate Metabolizers (IM), and Poor Metabolizers (PM) in
CNS disorders.
(C: Controls, N=315; ANX: Anxiety, N=285; DEP: Depression, N=419;
PSY: Psychosis, N=162; STR: Stroke, N=67; AD: Alzheimer’s disease,
N=231; PAR: Parkinson’s disease, N=73; ADHD: Attention Deficit
Hyperactivity Disorder, N=42; MIG: Migraine, N=217; EPI: Epilepsy,
N=71; VD: Vascular dementia, N=198; VE: Vascular encephalopathy,
N=380; MS: Multiple sclerosis, N=21; CVI: Cerebrovascular insufficiency,
N=138; BT: Brain tumor, N=11; CNN: Cranial nerve neuropathy, N=25;
MR: Mental retardation, N=115; PTBS: Post-traumatic brain syndrome,
N=59).
Significant differences were found between controls and patients with
MR (p<0.05), but not with other CNS disorders; however, patients with
ANX showed differences with respect to PSY (p<0.02), STR (p<0.02),
and MR (p<0.03). Other significant differences were found between
DEP and EPI (p<0.02), PSY and MS (p<0.04), and STR and MS
(p<0.05) [205].
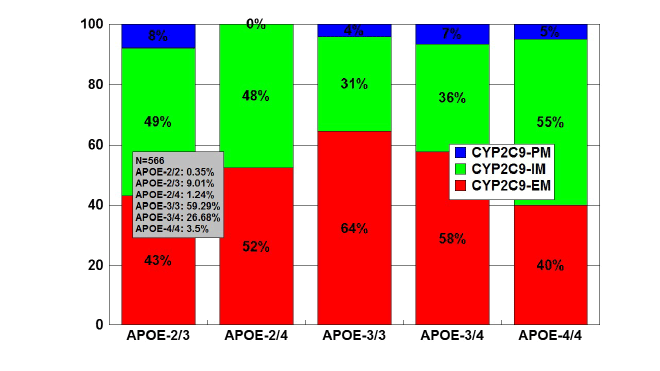
Figure 9: Distribution and frequency of CYP2C9 Extensive Metabolizers
(EM), Intermediate Metabolizers (IM), and Poor Metabolizers (PM)
associated with APOE genotypes in patients with Alzheimer’s disease.
Patients (N=566) were classified according to their APOE genotype
(APOE-2/2, 0.35%; APOE-2/3, 9.01%; APOE-2/4, 1.24%; APOE-3/3,
59.19%; APOE-3/4, 26.68%; APOE-4/4, 3.53%) and the distribution
and frequency of CYP2C9 variants were studied in each APOE-related
group. Significant differences were found between APOE-3/3 and
APOE-2/3 (p<0.001), and a different pattern of CYP2C9 variants was
also observed between APOE-3/3 and APOE-4/4 carriers (p<0.08).
CYP2C19
CYP2C19 is a gene (90.21 kb) with 9 exons mapped on 10q24.1q24.3.
RNA transcripts of 1901 bp, 2395 bp, and 1417 bp are expressed in liver
cells where a protein of 55.93 kDa (490 aa) is identified. Nearly 500 drugs
are CYP2C19-related, 281 acting as substrates (151 are major substrates,
130 are minor substrates), 263 as inhibitors (72 weak, 127 moderate,
and 64 strong inhibitors), and 23 as inducers of the CYP2C19 enzyme
[86]. About 541 SNPs have been detected in the CYP2C19 gene. The
frequencies of the 3 major CYP2C19 geno-phenotypes in the control
population are CYP2C19-*1/*1-EMs 68.54%, CYP2C19-*1/*2-IMs
30.05%, and CYP2C19-*2/*2-PMs 1.41%. EMs, IMs, and PMs account for
69.89%, 30.11%, and 0%, respectively, in AD, and 66.27%, 30.12%, and
3.61%, respectively, in VD [7] (Figure 11).
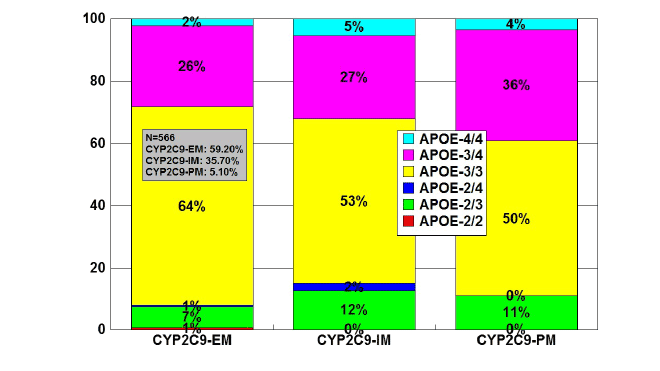
Figure 10: Distribution and frequency of APOE genotypes associated
with CYP2C9 Extensive Metabolizers (EM), Intermediate Metabolizers
(IM), and Poor Metabolizers (PM) in patients with Alzheimer’s disease.
Patients (N=566) were classified according to their CYP2C9 genophenotype
(EMs: 59.20%; IMs: 35.70%; PMs: 5.10%) and the
distribution and frequency of APOE genotypes were studied in each
CYP2C9-related group.
APOE-CYP2C19 association
The frequencies of CYP2C19-EMs, IMs, and PMs in a sample of 569
patients were 74.34%, 24.78%, and 0.88%, respectively. The distribution
of APOE-associated CYP2C19 geno-phenotypes was as follows: in APOE-
2/2 (N=2), CYP2C19-EMs 50%, and CYP2C19-IMs 50%; in APOE-2/3
(N=52), CYP2C19-EMs 65.43%, CYP2C19-IMs 34.62%; in APOE-2/4
(N=7), CYP2C19-EMs 71.43%, and CYP2C19-IMs 28.57%; in APOE-3/3
(N=336), CYP2C19-EMs 75.30%, CYP2C19-IMs 23.51%, and CYP2C19-
PMs 1.19%); in APOE-3/4 (N=152), CYP2C19-EMs 75.65%, CYP2C19-
IMs 23.03%, and CYP2C19-PMs 1.32%; and in APOE-4/4 (N=20),
CYP2C19-EMs 75%, and CYP2C19-IMs 25% (Figure 12). CYP2C19-PMs
are very rare among AD patients (60% APOE-3/3 and 40% APOE-3/4).
There is a small reduction in APOE-3/3 carriers among CYP2C19-IMs,
and a notable increase in APOE-3/4 carriers among CYP2C19-PMs
(Figure 13).
CYP3A4/5
CYP3A4 is a gene (27.2 kb) with 13 exons mapped on 7q21.1. RNA
transcripts of 2153 bp, 651 bp, 564 bp, 2318 bp and 2519 bp are expressed
in intestine, liver, prostate and other tissues where 4 protein variants of
57.34 kDa (503 aa), 17.29 kDa (153 aa), 40.39 kDa (353 aa), and 47.99 kDa
(420 aa) are identified. The human CYP3A locus contains the three CYP3A
genes (CYP3A4, CYP3A5 and CYP3A7), three pseudogenes, as well as a
novel CYP3A gene termed CYP3A43. The gene encodes a putative protein
with between 71.5% and 75.8% identity to the other CYP3A proteins.
The predominant hepatic form is CYP3A4, but CYP3A5 contributes
significantly to the total liver CYP3A activity. This enzyme metabolizes
over 1,900 drugs, 1,033 acting as substrates (897 are major substrates, 136
are minor substrates), 696 as inhibitors (118 weak, 437 moderate, and
141 strong inhibitors), and 241 as inducers of the CYP3A4 enzyme [86].
About 347 SNPs have been identified in the CYP3A4 gene (CYP3A4*1A:
Wild-type), 25 of which are of clinical relevance. Concerning CYP3A4/5
polymorphisms in AD, 82.75% of the cases are EMs (CYP3A5*3/*3),
15.88% are IMs (CYP3A5*1/*3), and 1.37% are UMs (CYP3A5*1/*1).
Unlike other human P450s (CYP2D6, CYP2C19) there is no evidence of a
‘null’ allele for CYP3A4 [86].
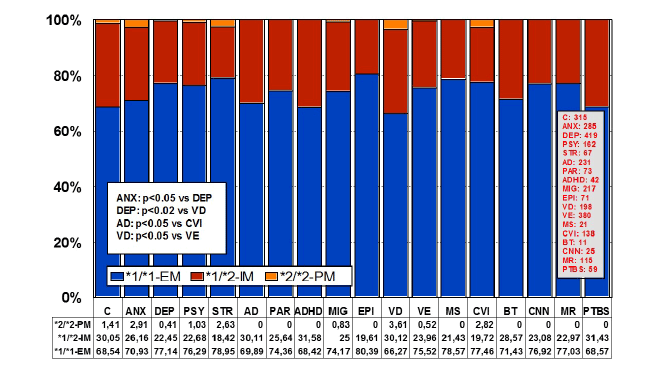
Figure 11: Distribution and frequency of CYP2C19 Extensive
Metabolizers (EM), Intermediate Metabolizers (IM), and Poor
Metabolizers (PM) in CNS disorders.
(C: Controls, N=315; ANX: Anxiety, N=285; DEP: Depression, N=419;
PSY: Psychosis, N=162; STR: Stroke, N=67; AD: Alzheimer’s disease,
N=231; PAR: Parkinson’s disease, N=73; ADHD: Attention Deficit
Hyperactivity Disorder, N=42; MIG: Migraine, N=217; EPI: Epilepsy, N=71;
VD: Vascular dementia, N=198; VE: Vascular encephalopathy, N=380; MS:
Multiple sclerosis, N=21; CVI: Cerebrovascular insufficiency, N=138; BT:
Brain tumor, N=11; CNN: Cranial nerve neuropathy, N=25; MR: Mental
retardation, N=115; PTBS: Post-traumatic brain syndrome, N=59).
No significant differences between controls and patients with CNS
disorders were found; however, differences were found between ANX
and DEP (p<0.05), DEP and VD (p<0.02), AD and CVI (p<0.05), and
VD and VE (p<0.05) [205].
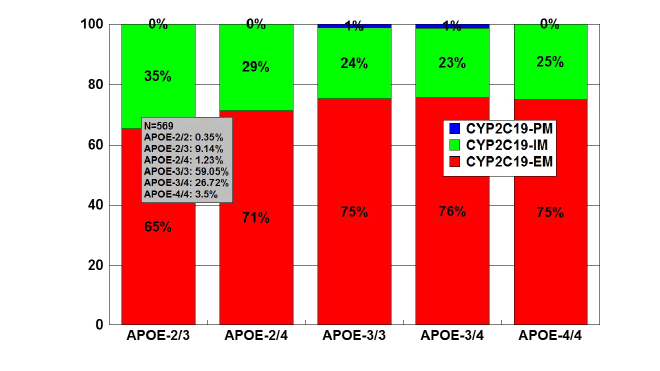
Figure 12: Distribution and frequency of CYP2C19 Extensive
Metabolizers (EM), Intermediate Metabolizers (IM), and Poor
Metabolizers (PM) associated with APOE genotypes in patients with
Alzheimer’s disease.
Patients (N=569) were classified according to their APOE genotype
(APOE-2/2,0.35%; APOE-2/3, 9.14%; APOE-2/4, 1.23%; APOE-3/3,
59.05%; APOE-3/4, 26.72%; APOE-4/4, 3.51%) and the distribution
and frequency of CYP2C19 geno-phenotypes were studied in each
APOE-related group.
APOE-CYP3A4/5 interaction
In a series of 347 AD cases, 79.94% were found to be CYP3A5-EMs,
19.47% CYP3A4-IMs, and 0.59% CYP3A4-RM (rapid metabolizers). The
distribution of CYP3A5-EMs and IMs was very similar among APOE
genotypes, except in APOE-2/4 carriers where the presence of IMs was
twice higher than in carriers of the other genotypes (Figure 14). Only 2
cases of CYP3A4-RMs were found exclusively associated with APOE-3/3
genotypes (Figure 15).
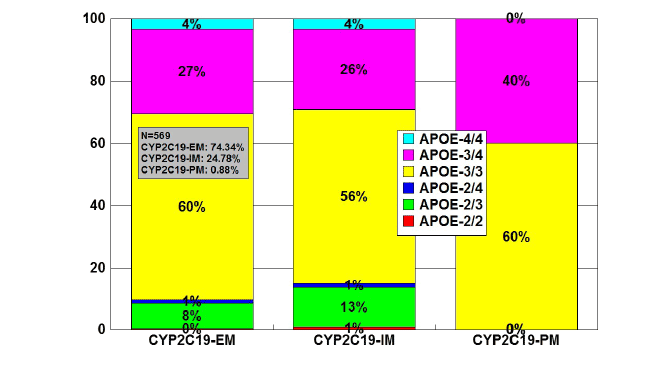
Figure 13: Distribution and frequency of APOE genotypes associated
with CYP2C19 Extensive Metabolizers (EM), Intermediate Metabolizers
(IM), and Poor Metabolizers (PM) in patients with Alzheimer’s disease.
Patients (N=569) were classified according to their CYP2C19 genophenotypes
(CYP2C19-EM, 74.34%; CYP2C19-IM, 24.78%; CYP2C19-
PM, 0.88%) and the distribution and frequency of APOE genotypes
were studied in each CYP2C19-related group.
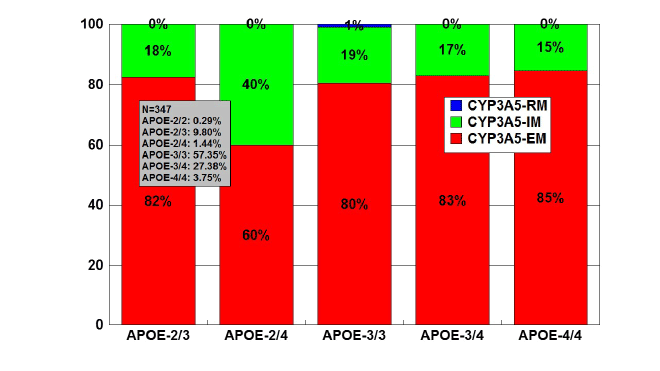
Figure 14: Distribution and frequency of CYP3A5 Extensive Metabolizers
(EM), Intermediate Metabolizers (IM), and Rapid Metabolizers (RM)
associated with APOE genotypes in patients with Alzheimer’s disease.
Patients (N=347) were classified according to their APOE genotype
(APOE-2/2, 0.29%; APOE-2/3, 9.80%; APOE-2/4, 1.44%; APOE-3/3,
57.35%; APOE-3/4, 27.38%; APOE-4/4, 3.75%) and the distribution and
frequency of CYP3A5 variants were studied in each APOE-related group.
CYP clustering
The construction of a genetic map integrating the most prevalent
CYP2D6+CYP2C19+CYP2C9 polymorphic variants in a trigenic cluster
yields 82 different haplotype-like profiles. The most frequent trigenic
genotypes in the AD population are *1*1-*1*1-*1*1 (25.70%), *1*1-*1*2-
*1*2 (10.66%), *1*1-*1*2-*1*1 (10.45%), *1*4-*1*1-*1*1 (8.09%), *1*4-
*1*2-*1*1 (4.91%), *1*4-*1*1-*1*2 (4.65%), and *1*1-*1*3-*1*3 (4.33%).
These 82 trigenic genotypes represent 36 different pharmacogenetic
phenotypes. According to these trigenic clusters, only 26.51% of the
patients show a pure 3EM phenotype, 15.29% are 2EM1IM, 2.04% are
pure 3IM, 0% are pure 3PM, and 0% are 1UM2PM (the worst possible
phenotype). This implies that only one-quarter of the population
processes normally the drugs which are metabolized via CYP2D6,
CYP2C9 and CYP2C19 (approximately 60% of the drugs of current use)
[26]. Taking into consideration the data available, it might be inferred
that at least 20-30% of the AD population may exhibit an abnormal
metabolism of cholinesterase inhibitors and/or other drugs which
undergo oxidation via CYP2D6-related enzymes. Approximately 50% of
this population cluster would show an ultrarapid metabolism, requiring
higher doses of cholinesterase inhibitors in order to reach a therapeutic
threshold, whereas the other 50% of the cluster would exhibit a poor
metabolism, displaying potential adverse events at low doses. If we take
into account that approximately 60-70% of therapeutic outcomes depend
upon pharmacogenomic criteria (e.g. pathogenic mechanisms associated
with AD-related genes), it may be postulated that pharmacogenetic and
pharmacogenomic factors are responsible for 75-85% of the therapeutic
response (efficacy) in AD patients treated with conventional drugs
[1,7,21,22,24-27,34-38,94].
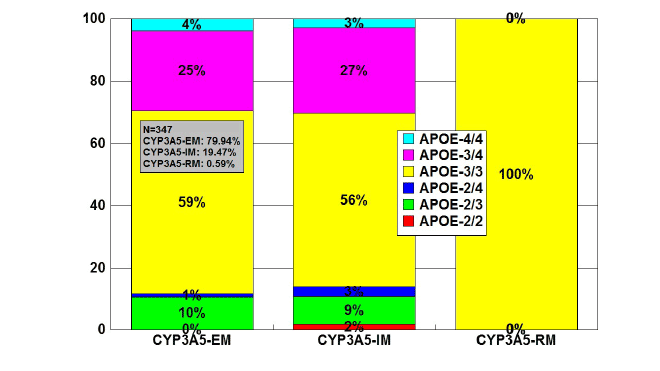
Figure 15: Distribution and frequency of APOE genotypes associated
with CYP3A5 Extensive Metabolizers (EM), Intermediate Metabolizers
(IM), and Rapid Metabolizers (RM) in patients with Alzheimer’s disease.
Patients (N=347) were classified according to their CYP3A5 genophenotype
(CYP3A5-EM, 79.94%; CYP3A5-IM, 19.47%; CYP3A5-RM,
0.59%) and the distribution and frequency of APOE genotypes were
studied in each CYP3A5-related group.
Genes encoding drug transporters
ABC genes, especially ABCB1 (ATP-binding cassette, subfamily B,
member 1; P-glycoprotein-1, P-gp1; Multidrug Resistance 1, MDR1)
(7q21.12), ABCC1 (9q31.1), ABCG2 (White1) (21q22.3), and other genes
of this family encode proteins which are essential for drug metabolism
and transport. The multidrug efflux transporters P-gp, MultidrugResistance
Associated Protein 4 (MRP4) and Breast Cancer Resistance
Protein (BCRP), located on endothelial cells lining brain vasculature,
play important roles in limiting movement of substances into and
enhancing their efflux from the brain. Transporters also cooperate with
Phase I/Phase II metabolism enzymes by eliminating drug metabolites.
Their major features are their capacity to recognize drugs belonging to
unrelated pharmacological classes, and their redundancy, by which a
single molecule can act as a substrate for different transporters. This
ensures an efficient neuroprotection against xenobiotic invasions. The
pharmacological induction of ABC gene expression is a mechanism
of drug interaction, which may affect substrates of the up-regulated
transporter, and overexpression of MDR transporters confers resistance
to anticancer agents and CNS drugs [95,96].
Aberrant cholesterol trafficking and accumulation may contribute
to the early onset of AD. Several ATP-Binding Cassette (ABC)
transporters, such as ABCA1, ABCG1, ABCG5, and ABCG8 have been
shown to play important roles in the regulation of cellular cholesterol
homeostasis by mediating cholesterol efflux. Mutations in ABC
transporters influence pathogenesis and therapeutics of brain disorders
[97].
Genome-wide significance in fully adjusted models was observed for
a SNP in ABCA7 (rs115550680, allele = G; frequency, 0.09 cases and 0.06
controls), which is in linkage disequilibrium with SNPs associated with
AD in Europeans. The effect size for the SNP in ABCA7 was comparable
with that of the APOEє4-determining SNP rs429358 (allele = C; frequency,
0.30 cases and 0.18 controls) [98].
ABCB1
ABCB1(ATP-binding cassette, sub-family B (MDR/TAP), member
1; Doxorubicin resistance; Multidrug resistance 1; Multidrug resistance
protein 1; P glycoprotein 1; P glycoprotein 1/multiple drug resistance 1;
P-Glycoprotein 1; P-glycoprotein-1/multiple drug resistance-1; P-gp) is
probably the most important drug transporter in the brain. The ABCB1
gene maps on 7q21.12 spanning 209.39 kb (29 Exons) with the structure
of a P-glycoprotein and a Y-box sequence 5’-CTGATTGG-3’ in its cisregulatory
elements. Several transcripts/variants (ABCB1-001: 4645 bp;
ABCB1-002: 3602 bp; ABCB1-003: 461 bp; ABCB1-004: 582 bp; ABCB1-
005: 555 bp; ABCB1-006: 913 bp; ABCB1-007: 1864 bp; ABCB1-008:
642 bp; ABCB1-009: 787 bp; ABCB1-010: 539 bp; ABCB1-201: 345 bp)
are highly expressed in adrenal gland, Blood Brain Barrier (BBB), brain,
kidney, liver, placenta, small intestine and uterus, and low expression is
present in many other tissues. These transcripts encode a protein (ABCB1-
001: 141.48 kDa; 1280 aa. ABCB1-002: 5.89 kDa; 51 aa. ABCB1-003: 5.68
kDa; 48 aa. ABCB1-201: 2.52 kDa; 22 aa) of the ATP binding cassette
superfamily, subfamily B (MDR/TAP) with two ATP binding and two
transmembrane (2TM) domains (2 x 6 segments), acting as a transport
carrier and a lipid translocase of broad specificity.
This is a large transmembrane protein which is an integral part of the
BBB and functions as a drug-transport pump transporting a variety of
drugs from the brain back into the blood. Functions of this protein include
the following: ABC transporter, traffic ATPase, energy-dependent efflux
pump responsible for decreased drug accumulation in multidrug-resistant
cells; potentially implicated in cholesterol transport; may maintain neural
stem/progenitor cells in an undifferentiated state and could be a neural
stem/progenitor marker.
About 1630 ABCB1 variants have been identified [86]. Of interest,
ABCB1 has approximately 116 polymorphic sites in Caucasians and 127 in
African-Americans with a minor allele frequency greater than 5%. Some of
the most commonly studied variants are 1236C>T, 2677G>A/T and 3435C>T
and the most commonly studied haplotype involves the 1236, 2677 and 3435
(TTT) SNPs and 3 intronic SNPs (intron 9, intron 13, intron 14) named
ABCB1*13. There are many other ABCB1 variants such as -129C>T (5’-UTR),
61A>G (Asn21Asp) and 1199G>A (Ser400Asn) that have been studied in vivo
and in vitro. To date, there is no clear consensus on the impact of any of
these variants on drug disposition, response or toxicity.
Variants of the ABCB1 gene have been associated with a diverse
number of diseases and with a great variety of drugs, natural products
and endogenous agents [86]. Over 1,270 drugs have been reported to
be associated with the Abcb1 transporter protein (P-gp), of which 490
are substrates, 618 are inhibitors, 182 are inducers, and 269 additional
compounds which belong to different pharmacological categories of
products with potential Abcb1 interaction [86].
ATP-Binding Cassette (ABC) transporters, which are localized on the
surface of brain endothelial cells of the BBB and brain parenchyma, may
contribute to the pathogenesis of AD. ABC transporters including ABCB1
(P-glycoprotein, P-gp), ABCG2 (breast cancer resistant protein, BCRP),
ABCC1 (multidrug resistance protein 1, MRP1), and the cholesterol
transporter ABCA1 play important roles in the pathogenesis of AD and
Aβ peptide deposition inside the brain [99-104]. Decreased clearance of
Aβ from the brain may lead to elevated Aβ levels. One of the clearance
pathways of Aβ is transport across the BBB via efflux transporters.
P-glycoprotein, an efflux pump highly expressed at the endothelial cells of
the BBB, has been shown to transport Aβ. The P-glycoprotein transporter
at the BBB is compromised in AD, and decreased P-glycoprotein function
may be involved in the pathogenesis of AD [103].
In addition to the age-related decrease in P-gp expression, Aβ1-42 itself
downregulates the expression of P-gp and other Aβ-transporters, which
could exacerbate the intracerebral accumulation of Aβ and thereby
accelerate neurodegeneration in AD and cerebral β- Aβ angiopathy [102].
Furthermore, amyloid efflux transporter expression at the BBB declines
with aging in normal conditions [105], and expression of P-gp protein
is significantly lower in the hippocampal vessels of patients with AD
compared to normal individuals [106].
The Low-Density Lipoprotein Receptor-Related Protein-1 (LRP-1) and
the ATP-Binding Cassette (ABC) protein ABCB1 (P-glycoprotein) are
involved in the efflux of Aβ across the BBB. Other ABC proteins, such as
members of the G subfamily, are also involved in the BBB clearance of Aβ.
ABCG2 and ABCG4 mediate the cellular efflux of [3
H]Aβ1-40. Probucol
inhibits the efflux of [3
H]Aβ1-40 from HEK293-abcg4 cells. GF120918
(a dual inhibitor of Abcb1 and Abcg2) strongly enhances the uptake of
[3
H]Aβ1-40 by the brains of Abcb1-deficient mice, but not by the brains
of Abcb1/Abcg2-deficient mice, suggesting that Abcg2 is involved in the
transport of Aβ at the mouse BBB. Abcg4 acts in concert with Abcg2 to
efflux Aβ from the brain across the BBB [107].
ATP binding cassette subfamily G member 2 (ABCG2) is involved
in Aβ-β transport and was found to be up-regulated in AD brains. A
functional polymorphism of the ABCG2 gene (C421A; rs2231142)
(ABCG2 C/C genotype) was associated with AD in the Hungarian
population. The ABCG2 C/C genotype and the APOE-4 allele may also
exert an interactive effect on AD risk [108].
Single-nucleotide polymorphisms in the ABCB1 gene have been
associated with altered P-glycoprotein expression and function.
P-glycoprotein function at the BBB can be quantified in vivo using
the P-glycoprotein substrate tracer (R)-[11C] verapamil and Positron
Emission Tomography (PET). Three different kinds of imaging probes
have been described to measure ABC transporters in vivo: (i) radiolabeled
transporter substrates, (ii) radiolabeled transporter inhibitors, and (iii)
radiolabeled prodrugs which are enzymatically converted into transporter
substrates in the organ of interest [109]. Van Assema et al. [110] assessed
the effects of C1236T, G2677T/A and C3435T single-nucleotide
polymorphisms in ABCB1 on BBB P-glycoprotein function in healthy
subjects and patients with AD. In healthy controls, binding potential did
not differ between subjects without and with one or more T present in
C1236T, G2677T and C3435T. In contrast, patients with AD with one or
more T in C1236T, G2677T and C3435T had significantly higher binding
potential values than patients without a T. There was a relationship
between binding potential and T dose in C1236T and G2677T. In AD
patients, C1236T, G2677T/A and C3435T SNPs may be related to changes
in P-glycoprotein function at the BBB, and genetic variations in ABCB1
might contribute to the progression of Aβ-β deposition in the brain. Kohen
et al. [111] investigated a possible association between 2 common ABCB1
polymorphisms, G2677T/A (Ala893Ser/Thr) and C3435T, AD, and CSF
levels of Aβ, and no strong evidence for association was found. Frankfort
et al. [112] studied ABCB1 SNPs (C1236T in exon 12, G2677T/A in exon
21 and C3435T in exon 26) and inferred haplotypes in patients with
dementia and age-matched non-demented control patients and found
no differences between both groups; however, in a transcriptome analysis
of leukocytes from patients with mild cognitive impairment (MCI), AD,
as well as normal controls, only the ABCB1 gene exhibited significantly
positive correlation with MMSE scores, representing a novel biomarker
of AD [113].
Aβ transport (flux) across the BBB is thought to contribute to the
pathogenesis of AD and also the elimination of toxic amyloid from the
brain by immunotherapy. Several BBB transporters have been implicated
in Aβ exchange between brain parenchyma and the circulation, including
efflux transporters P-glycoprotein/ABCB1 and BCRP/ABCG2. Deficiency
of either of the two major efflux pumps, Abcb1 and Abcg2, implicated
in Aβ trafficking across the BBB, results in increased accumulation of
peripherally-injected Aβ1-40 in the brain [114].
The drug transporter ABCB1 directly transports Aβ from the brain
into the blood circulation, whereas the cholesterol transporter ABCA1
neutralizes Aβ aggregation capacity in an Apolipoprotein E (ApoE)-
dependent manner, facilitating subsequent Aβ elimination from the
brain [115]. Cascorbi et al. [116] genotyped selected variants in ABCA1,
ABCA7, ABCB1, ABCC2 and ABCG2 in DNAs from brain tissue of
71 AD cases with Consortium to Establish a Registry for Alzheimer’s
Disease (CERAD) neuropathological stages B/C and 81 controls. The
novel ABCA7 SNP, rs3752246, tended to be associated with AD. ABCB1
variants were significantly less frequent in AD cases older than 65 years of
age and among females. This association of ABCB1 2677G>T (rs2032582)
was more pronounced in APOE-4 negative cases. Only ABCC2 3972C>T
(rs3740066) was significantly associated with AD risk.
Efflux transporter P-glycoprotein (P-gp) at the BBB restricts
substrate compounds from entering the brain and may thus contribute
to pharmacoresistance in CNS disorders, cancer and brain infections.
Positron Emission Tomography (PET) has become a promising method
to study the role of P-gp at the BBB. The first PET study of P-gp function
was conducted in 1998, and over the past 15 years two main categories of
P-gp PET tracers have been investigated: tracers that are substrates of P-gp
efflux and tracers that are inhibitors of P-gp function [117].
The ABC transporter Pgp protects the brain from accumulation of
lipophilic compounds by active efflux transport across the BBB. Müllauer
et al. [118] investigated the suitability of the radiolabeled Pgp inhibitors
[11C] elacridar and [11C] tariquidar to visualize Pgp density in rat brain
with PET. The small Pgp binding signals obtained with [11C] elacridar and
[11C] tariquidar limit the applicability of these tracers to measure cerebral
Pgp density.
Molecular transporters that are expressed in brain, especially at the
BBB, are therapeutic targets in the treatment of AD. Some ATP-Binding
Cassette (ABC) transporters, particularly P-glycoprotein (ABCB1), MRP1
(ABCC1) and BCRP (ABCG2), have been implicated in the clearance of
neurotoxic polypeptides that characteristically accumulate in the brain,
such as Aβ peptides. A benzopyrane derivative with P-gp stimulating
properties has been proposed as a candidate agent to decrease Aβ
accumulation in AD [119]. Lipid transporters of the A-branch of ABC
transporters are also potentially involved in AD pathogenesis. Induction of
transporters via the activation of specific nuclear receptors may represent
a novel approach to restoring diminished BBB function. Transporters in
the brain capillary endothelium regulate the permeation of therapeutic
compounds into the brain [120,121].
Induction of the multidrug resistance protein 1 (MDR1)/Pglycoprotein
(P-gp) by the Vitamin D Receptor (VDR) was investigated
in isolated rat brain capillaries and rat (RBE4) and human (hCMEC/
D3) brain microvessel endothelial cell lines. Incubation of isolated rat
brain capillaries with the VDR ligand, 1α,25-dihydroxyvitamin D3
[1,25OH2 D3 ] increased P-gp protein expression fourfold. Incubation with 1,25OH2 D3 increased P-gp transport activity by 25-30%. In RBE4 cells, Mdr1b mRNA was induced in a concentration-dependent manner by exposure to 1,25OH2 D3. Concomitantly, P-gp protein expression increased 2.5-fold and was accompanied by a 20-35% reduction in cellular
accumulation of the P-gp substrates, rhodamine 6G (R6G), and HiLyte
Fluor 488-labeled human amyloid-β 1-42 (hAβ42). In hCMEC/D3 cells,
exposure to 1,25OH2
D3
increased MDR1 mRNA expression (40%) and
P-gp protein; and reduced cellular accumulation of R6G and hAβ42 by
30%. VDR activation up-regulates Mdr1/MDR1 and P-gp protein in brain
capillaries and microvascular endothelia, implicating a role for VDR in
increasing the brain clearance of P-gp substrates, including hAβ42 in AD
[122].
Since P-gp prevents the entry of compounds into the brain by an
active efflux mechanism at the BBB, inhibition of P-gp may help to
enhance drug penetration. New reversible inhibitors of P-gp have been
developed. Some galantamine-like compounds inhibit the efflux of the
fluorescent P-gp substrate rhodamine 123 in cancer cells that over-express
P-gp, and also inhibit the efflux of therapeutic substrates of P-gp, such
as doxorubicin, daunomycin and verapamil. These compounds modulate
P-gp-mediated efflux by competing for the substrate binding sites [123].
Activation of the Liver X Receptors (LXRs) by natural or synthetic
agonists decreases the amyloid burden and enhances cognitive function in
transgenic murine models of AD. LXR activation may affect the transport
of Aβ peptides across the BBB. LXR agonists (24S-hydroxycholesterol,
27-hydroxycholesterol and T0901317) modulate the expression of target
genes involved in cholesterol homeostasis (ABCA1) and promote cellular
cholesterol efflux to apolipoprotein A-I and high density lipoproteins.
LXR stimulation increases the expression of the ABCB1 transporter,
which restricts Aβ peptide influx [124].
It is also important that drugs for AD treatment optimize CNS
penetration by minimizing hydrogen bond donors and reducing P-gpmediated
efflux [125-127]. The increase of P-glycoprotein expression and
activity by a P-gp inducer could be an effective pharmacological strategy
in slowing or halting the progression of AD. A decrease of approximately
10-35% in 124I-Aβ1-40 intracellular accumulation was observed in cells
treated with rifampicin, dexamethasone, caffeine, verapamil, hyperforin,
β-estradiol and pentylenetetrazole (P-gp inducers) [128]. Perrone et al.
[129] validated the new dye-probe β-amyloid (1-40) HiLyte Fluor™ TRlabeled
(Ab-HiLyte) (Anaspec) P-gp-mediated transport in the ex vivo rat
everted gut sac assay by using MC18 or MC266, a fully characterized P-gp
inhibitor and substrate, respectively, and compared it with the commonlyused
dye rhodamine, demonstrating that the new dye probe, Ab-HiLyte,
could be a probe of choice to unequivocally distinguish between a P-gp
substrate and an inhibitor.
Mehta et al. [121] assessed the impact of AD-associated BBB alterations
on the uptake of therapeutics into the brain of triple transgenic (3×TG)
AD mice. The brain uptake of passively diffusing markers, [3
H] diazepam
and [3
H] propranolol, decreased 54-60% in 3×TG mice, consistent with a
33.5% increase in the thickness of the cerebrovascular basement membrane
in 3×TG mice. Despite a 42.4% reduction in P-gp expression in isolated
brain microvessels from a sub-population of 3×TG mice, the brain uptake
of P-gp substrates ([3
H] digoxin, [3
H] loperamide and [3
H] verapamil) was
not different between genotypes, likely due to a compensatory thickening
in the cerebrovascular basement membrane counteracting any reduced
efflux of these lipophilic substrates.
Other transporters
Also of importance for CNS pharmacogenomics are transporters
encoded by genes of the solute carrier superfamily (SLC) and solute carrier
organic (SLCO) transporter family, responsible for the transport of multiple
endogenous and exogenous compounds, including folate (SLC19A1), urea
(SLC14A1, SLC14A2), monoamines (SLC29A4, SLC22A3), aminoacids
(SLC1A5, SLC3A1, SLC7A3, SLC7A9, SLC38A1, SLC38A4, SLC38A5,
SLC38A7, SLC43A2, SLC45A1), nucleotides (SLC29A2, SLC29A3),
fatty acids (SLC27A1-6), neurotransmitters (SLC6A2 (noradrenaline
transporter), SLC6A3 (dopamine transporter), SLC6A4 (serotonin
transporter, SERT), SLC6A5, SLC6A6, SLC6A9, SLC6A11, SLC6A12,
SLC6A14, SLC6A15, SLC6A16, SLC6A17, SLC6A18, SLC6A19), glutamate
(SLC1A6, SLC1A7), and others [130]. Some Organic Anion Transporters
(OAT), which belong to the Solute Carrier (SLC) 22A family, are also
expressed at the BBB, and regulate the excretion of endogenous and
exogenous organic anions and cations [131]. The transport of amino acids
and di- and tripeptides is mediated by a number of different transporter
families, and the bulk of oligopeptide transport is attributable to the
activity of members of the SLC15A superfamily (Peptide Transporters 1
and 2 [SLC15A1 (PepT1) and SLC15A2 (PepT2)], and Peptide/Histidine
Transporters 1 and 2 [SLC15A4 (PHT1) and SLC15A3 (PHT2)]). ABC
and SLC transporters expressed at the BBB may cooperate to regulate the
passage of different molecules into the brain [132]. Polymorphic variants
in ABC and SLC genes may also be associated with pathogenic events
in CNS disorders and drug-related safety and efficacy complications
[8,130]. For instance, an important issue to be elucidated is the role of
transporters in patients under chronic treatment with psychotropic drugs
or exposed to general anesthesia. Chen et al. [133] studied the potential
influence of Endotracheal Tube Intubation General Anesthesia (ETGA),
Intravenous Injection General Anesthesia (IVGA) or Intramuscular
Injection General Anesthesia (IMGA), and heavy sedation on dementia
in Taiwan and found that individuals exposed to surgery under ETGA
and those exposed to surgery under IVGA or IMGA were at significantly
higher risk of dementia in a dose-response relationship, whereas surgery
under heavy sedation was not associated with increased risk of dementia.
Subjects who had received surgery under ETGA with comorbidities
such as stroke, hypertension, diabetes mellitus, and atherosclerosis could
have a potential relationship with dementia risk [32]. Interestingly, the
anesthetics propofol and thiopental are associated with Αβ assembly and
GM1 expression on the neuronal cell surface through the γ-aminobutyric
acid A receptor, and both compounds have direct and indirect inhibitory
effects on Αβ fibrillogenesis [134].
Pharmacogenomics of anti-dementia drugs
Donepezil
Donepezil is a centrally active, reversible acetylcholinesterase inhibitor
which increases the acetylcholine available for synaptic transmission in the
CNS. The therapeutic response of donepezil is influenced by pathogenic
gene variants (APOE, CHAT), as well as mechanistic gene polymorphic
variants of CHAT, ACHE, and BCHE. Donepezil is a major substrate of
CYP2D6, CYP3A4, ACHE, and UGTs, inhibits ACHE and BCHE, and is
transported by ABCB1 [8,21,24,25,28,35,135-137] (Table 1). Most studies
convey that CYP2D6 variants affect donepezil efficacy and safety in AD
[8,21,24,25,35,86,135-137]. The common variant rs1080985 of CYP2D6
was found to be associated with poor response to donepezil [138,139].
A high-throughput genetic analysis of CYP2D6 polymorphisms
discriminated responders/non-responders of the CYP2D6 allele *2A. A
higher frequency of mutated alleles was observed in responder than in
non-responder patients (75.38% vs. 43.48%). The presence of a mutated
allele of CYP2D6 was associated with a response to CYP2D6-metabolized
drugs [140]. In agreement with this criterion, in an Italian study 67% of
patients were responders and 33% were non-responders to donepezil
treatment. A significantly higher frequency of gene variants conferring
decreased or absent enzyme activity was observed in responder than
in non-responder patients (73.68% vs. 36.84%) [141]. Among Chinese
patients, 58.3% were responders and 41.7% were non-responders to
donepezil treatment. AD patients with the mutant allele CYP2D6*10 may
respond better to donepezil than those with the wild allele CYP2D6*1. A
significantly higher frequency of patients with genotypes CYP2D6*1/*10
and *10/*10 were found in responders than in non-responders. Patients
with CYP2D6*1/*10 and *10/*10 genotypes had higher steady-state
plasma concentrations of donepezil and improved cognition scores than
those with the CYP2D6*1/*1 genotype [142]. However, in other studies,
CYP2D6-PMs and UMs tend to be poor responders to conventional doses
of donepezil as compared to EMs and IMs [1,7,21,22,24-28,34-38,94,143-
145]. In contrast, a Polish group could not find any influence of the
rs1080985 SNP on response to treatment with donepezil in AD [146].
Magliulo et al. [147] evaluated the impact of CYP3A4 (*1B, *3, and
*4), CYP3A5 (*2, *3, and *6), and ABCB1 (3435C>T, 2677G>T/A, and
1236C>T) polymorphisms on donepezil disposition and clinical outcome
in 54 Italian AD patients. Three patients carried one detrimental CYP3A4
allelic variant, and 12 carried one functional CYP3A5*1 allele. No
association was found between CYP3A4 or CYP3A5 genotypes and plasma
donepezil concentrations, or between genotypes and clinical response.
The most common ABCB1 haplotypes were 1236C/2677G/3435C (46%)
and 1236T/2677T/3435T (41%). Patients homozygous for the T/T/T
haplotype had lower plasma donepezil concentration-to-dose ratios and
better clinical response than patients with other genotypes.
Galantamine
Galantamine is a reversible and competitive acetylcholinesterase
inhibitor leading to an increased concentration of acetylcholine at
cholinergic synapses. This drug also modulates nicotinic acetylcholine
receptors and may increase glutamate and serotonin levels. APOE, APP,
ACHE, BCHE, CHRNA4, CHRNA7, CHRNB2 variants may potentially
influence galantamine efficacy and safety; it is a major substrate of
CYP2D6, CYP3A4, and UGT1A1, and an inhibitor of ACHE and
BCHE [86,136,137,148-150] (Table 1). Major metabolic pathways are
glucuronidation, O-demethylation, N-demethylation, N-oxidation, and
epimerization. In extensive metabolizers for CYP2D6, urinary metabolites
resulting from O-demethylation represented 33.2% of the dose compared
with 5.2% in poor metabolizers, which showed correspondingly higher
urinary excretion of unchanged galantamine and its N-oxide. The
glucuronide of O-desmethyl-galantamine represented up to 19% of the
plasma radioactivity in extensive metabolizers but could not be detected
in poor metabolizers [151]. Galantamine is extensively metabolized by the
enzymes CYP2D6 and CYP3A and is a substrate of the P-glycoprotein.
Noetzli et al. [152] studied the relationship between genetic variants of
CYP2D6, CYP3A4/5 and ABCB1 with galantamine steady state plasma
concentrations. The CYP2D6 genotype seemed to be an important
determinant of galantamine pharmacokinetics, with CYP2D6 poor
metabolizers presenting 45% and 61% higher dose-adjusted galantamine
plasma concentrations than heterozygous and homozygous CYP2D6
extensive metabolizers.
However, Clarke et al. [153] were unable to make inferences
about an association between CYP2D6 phenotype and galantamine
responsiveness.The bioavailability of galantamine is increased by
co-administration with paroxetine, ketoconazole and erythromycin
[154]. In healthy subjects and in AD patients, the co-administration
of galantamine with ketoconazole (a CYP3A4 strong inhibitor)
or paroxetine (a CYP2D6 strong inhibitor) leads to a 30% and
40% increase, respectively, in galantamine exposure compared to
galantamine given alone [155]. Galantamine can interact with foods
which might alter its bioavailability and therapeutic effects. Zhai and Lu
[156] reported interaction between galantamine and capsaicin (trans-
8-methyl-N-vanillyl-6-nonenamide, CAP), a naturally-occurring
alkaloid extracted from the fruit of Capsicum plant family, which is
a common ingredient in spicy foods. The pretreatment of rats with
capsaicin resulted in a decrease in the AUC0-∞ of galantamine of about
49.70% compared with the control group. After oral administration
of galantamine (10 mg/kg), the apparent oral clearance of galantamine
was raised by 2.05-fold by pretreatment with capsaicin, indicating that the
chronic ingestion of high doses of capsaicin decreases the bioavailability
of galantamine, at least in rats.
Rivastigmine
Rivastigmine is a cholinesterase inhibitor which increases acetylcholine
in CNS through reversible inhibition of its hydrolysis by cholinesterase.
APOE, APP, CHAT, ACHE, BCHE, CHRNA4, CHRNB2 and MAPT
variants may affect its pharmacokinetics and pharmacodynamics. The
hepatic cytochrome P-450 (CYP-450) system is not involved in the
metabolism of rivastigmine [86,136,137,154,157] (Table 1). Sonali et al.
[158] studied the clinical effectiveness of CYP2D6, CYP3A4, CYP2C9/19,
and UGT polymorphisms on the steady-state plasma concentrations and
therapeutic outcome of rivastigmine monotherapy and combination
therapy in patients with AD in India. A significant allele frequency was
observed for the CYP2D6*3 polymorphism in patients under rivastigmine
combination therapy (A>del: 0.50 AD/0.20 controls), UGT2B7 (T: 0.17
AD/0.33 C), and UGT1A9*5 (A = 0.58 AD/0.26 C). Poor metabolizer
subjects of the UGT2B7 polymorphism in patients under rivastigmine
combination therapy have higher drug levels with a poor response to
treatment.
Tacrine
Tacrine was the first FDA-approved anti-dementia drug. Its use was
stopped due to hepatotoxicity. Tacrine is a cholinesterase inhibitor
which elevates acetylcholine in cerebral cortex by slowing degradation
of acetylcholine. ACHE, BCHE, CHRNA4, CHRNB2, APOE, MTHFR,
CES1, LEPR, GSTM1, and GSTT1 variants may affect its therapeutic and
toxic effects. Tacrine is a major substrate of CYP1A2 and CYP3A4, a
minor substrate of CYP2D6, and is transported via SCN1A. Tacrine is
an inhibitor of ACHE, BCHE, and CYP1A2 [86] (Table 1). Both tacrine
and some tacrine-hybrids may cause an induction of CYP1A1, 2B1 and
3A2 expression [159]. Tacrine is associated with transaminase elevation
in up to 50% of patients. The mechanism of tacrine-induced liver damage
is influenced by genetic factors. The strongest association was found
between alanine aminotransferase levels and three SNPs within ATPbinding
cassette, subfamily B (MDR/TAP), member 4 (ABCB4) [160].
Memantine
Memantine is an NMDA receptor antagonist which binds preferentially
to NMDA receptor-operated cation channels; it may act by blocking
actions of glutamate, mediated in part by NMDA receptors, and it is also
an antagonist of GRIN2A, GRIN2B, GRIN3A, HTR3A and CHRFAM7A.
Several pathogenic (APOE, PSEN1, MAPT) and mechanistic gene variants
(GRIN2A, GRIN2B, GRIN3A, HTR3A, CHRFAM7A, c-Fos, Homer1b and
PSD-95) may influence its therapeutic effects. Memantine is a strong
inhibitor of CYP2B6 and CYP2D6, and a weak inhibitor of CYP1A2,
CYP2A6, CYP2C9, CYP2C19, CYP2E1, and CYP3A4 [86,137,161] (Table
1). Memantine is beneficial for AD patients in terms of cognition and in
the clinician’s global impression; however, some memantine-related major
side-effects (somnolence, weight gain, confusion, hypertension, nervous
system disorders, falling) [161] might be associated with pharmacogenetic
factors. Micuda et al. [162] studied the drug interaction potential of
memantine by elucidation of its inhibitory effects on cytochrome P450
enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6,
CYP2E1 and CYP3A4) using pooled Human Liver Microsomes (HLM)
and recombinant P450s. In HLM, memantine inhibited CYP2B6 and
CYP2D6 activities, showed no appreciable effect on CYP1A2, CYP2E1,
CYP2C9, or CYP3A4 activities, and decreased CYP2A6 and CYP2C19
activities. When co-administered with CYP2B6 substrates, a decrease in
metabolism of over 65% can be expected. Noetzli et al. [163] investigated
clinical and genetic factors influencing memantine disposition. A
population pharmacokinetic study was performed including data from
108 patients recruited in a naturalistic setting. Patients were genotyped
for common polymorphisms in renal cation transporters (SLC22A1/2/5,
SLC47A1, ABCB1) and nuclear receptors (NR1I2, NR1I3, RXR, PPAR)
involved in transporter expression. The average clearance was 5.2 L/h
with a 27% inter-individual variability. Glomerular filtration rate and
sex influenced memantine clearance. NR1I2 rs1523130 was identified as
the unique significant genetic covariate for memantine clearance, with
carriers of the NR1I2 rs1523130 CT/TT genotypes presenting a 16%
slower memantine elimination than carriers of the CC genotype.
Administration of NMDA receptor antagonists, such as ketamine
and MK-801, may induce psychotic-like behaviors, and ketamine can
exacerbate psychotic symptoms in patients with schizophrenia; in
contrast, memantine, a non-competitive NMDA receptor antagonist
approved for AD, may potentially display antipsychotic effects. The
molecular mechanisms by which these NMDA receptor antagonists cause
different neurochemical, behavioral, and clinical effects are associated with
differential expression of particular genes (Homer1a/Homer1b/PSD-95
signaling network), involved in glutamate-dependent synaptic plasticity,
as well as in psychosis pathophysiology and treatment. Ketamine and MK-
801 significantly induced the transcripts of immediate-early genes (Arc,
c-fos, and Homer1a) in cortical regions, whereas they reduced Homer1b
and PSD-95 expression in cortical and striatal regions. Memantine did not
increase Homer1a signal, whereas it induced c-fos in the somatosensory
and in the medial agranular cortices, not affecting Homer1b and PSD-
95 expression. When compared to ketamine and MK-801, memantine
significantly increased the expression of c-fos, Homer1b and PSD-
95. Overall, ketamine and MK-801 prominently increased Homer1a/
Homer1b expression ratio, whereas memantine elicited the opposite effect.
According to de Bartolomeis et al. [164], these data may support the view
that ketamine, MK-801 and memantine exert divergent effects on PSD
transcripts, which may contribute to their partially different behavioral
and clinical effects.
Martinelli-Boneschi et al. [165] conducted a genome-wide association
study in a cohort of 176 Italian AD patients treated with cholinesterase
inhibitors, classifying the patients into responders (positive, stable, or ≤1
worsening of MMSE score) and non-responders (>3 points worsening
in MMSE score) during a median follow-up of 0.85 years of treatment.
Among the 48 SNPs screened, only 2 SNPs were associated with response
to treatment: rs6720975A, and rs17798800A, an intergenic variant
potentially acting as a cis-regulator of neurobeachin (NBEA), an A
kinase-anchoring protein playing a substantial role in the maturation of
the nervous system.
Epigenetics
Epigenetics refers to phenotypic changes with no apparent alterations
in structural DNA. Classical epigenetic mechanisms, including DNA
methylation and histone modifications, and regulation by microRNAs
(miRNAs), are among the major regulatory elements that control
metabolic pathways at the molecular level, with epigenetic modifications
regulating gene expression transcriptionally and miRNAs suppressing
gene expression post-transcriptionally [166].
Vertebrate genomes undergo epigenetic reprogramming during
development and disease. Stable transmission of DNA methylation,
transcriptomes and phenotypes from parent to clonal offspring are
demonstrated in various asexual species, and clonal genotypes from
natural populations show habitat-specific DNA methylation [167].
Methylation varies spatially across the genome with a majority of
the methylated sites mapping to intragenic regions [168]. Not only
nuclear DNA (nDNA), but also mitochondrial DNA (mtDNA) may be
subjected to epigenetic modifications related to disease development,
environmental exposure, drug treatment and aging. mtDNA
methylation is attracting increasing attention as a potential biomarker
for the detection and diagnosis of diseases and the understanding of
cellular behavior [169].
Epigenetic mechanisms and miRNAs have recently been shown to
closely interact with each other, thereby creating reciprocal regulatory
circuits, which appear to be disrupted in AD [170]. Brain hypoperfusionrelated
changes in DNA methylation may also contribute to accelerate
neuronal death. Short-term, sub-lethal hypoxia results in long-lasting
changes to genome-wide DNA methylation status, and some of these
changes can be highly correlated with transcriptional modulation in a
number of genes involved in functional pathways [171].
Memory decline is a seminal symptom in dementia. Gene expression is
required for long-lasting forms of memory. Epigenetic mechanisms do not
only provide complexity in the protein regulatory complexes that control
coordinate transcription for specific cell function, but the epigenome
encodes critical information that integrates experience and cellular history
for specific cell functions as well. Epigenetic mechanisms provide a unique
mechanism of gene expression regulation for memory processes. Negative
regulators of gene expression, such as HDACs, have powerful effects on
the formation and persistence of memory. HDAC inhibition transforms a
subthreshold learning event into robust long-term memory and generates
a form of long-term memory that persists beyond the point at which
normal long-term memory fails [172]. Whereas increments in histone
acetylation have consistently been shown to favor learning and memory,
a lack thereof has been causally implicated in cognitive impairments in
neurodevelopmental disorders, neurodegeneration and aging. As histone
acetylation and cognitive functions can be pharmacologically restored
by histone deacetylase inhibitors, this epigenetic modification might
constitute a molecular memory aid on the chromatin and, by extension, a
new template for therapeutic interventions against cognitive decline [173].
Neurons, due to their post-mitotic state, high metabolism, and
longevity are particularly prone to the accumulation of DNA lesions.
DNA damage has been suggested as a major contributor to both ageassociated
neurodegenerative diseases and acute neurological injury. The
DNA damage response is a key factor in maintaining genome integrity.
It relies on highly dynamic post-translational modifications of the
chromatin and DNA repair proteins to allow signaling, access, and repair
of the lesion [174]. The repair of DNA lesions, particularly oxidative DNA
lesions, might be altered in AD. DNA damage is paralleled by a decrease
in DNA repair activities. DNA repair proteins might be inactivated
by oxidative induced post-translational modifications or degradation.
Activation of DNA repair pathways might generate death signals ending
with neuronal apoptosis. A link between environment-induced epigenetic
modification, oxidation, and repair of AD-related genes has been
proposed [175]. Early life exposure of rodents and primates to xenobiotics
may enhance the expression of genes associated with AD, repress the
expression of others, and increase the burden of oxidative DNA damage
in the aged brain. Epigenetic mechanisms that control gene expression
and promote the accumulation of oxidative DNA damage are mediated
through alterations in the methylation or oxidation of CpG dinucleotides.
Environmental influences occurring during brain development inhibit
DNA-methyltransferases, thus hypomethylating promoters of genes
associated with AD, such as APP. This early life imprint may sustain and
trigger later in life to increase the levels of APP and Aβ. Increased Aβ levels
promote the production of reactive oxygen species, which damage DNA
and accelerate neurodegenerative events. These early life perturbations
may result in hypomethylation as well as hypermethylation of genes.
The hypermethylated genes are rendered susceptible to Aβ-enhanced
oxidative DNA damage because methylcytosines restrict repair of adjacent
hydroxyguanosines [176]. Many AD-related genes contain methylated
CpG sites in their promoter regions, and a genome-wide decrease in
DNA methylation has been reported in AD [2,4,5,177,178]. A small bulk
of recent information [173,179,180] suggests that histone modifications
are present in AD: (i) histone acetylation is reduced in AD brain tissues
[181] and in AD transgenic models [173]; (ii) levels of HDAC6, a tau-
interacting protein and a potential modulator of tau phosphorylation
and accumulation, are increased in cortical and hippocampal regions
in AD [182]; mice lacking HDAC6 are cognitively normal, but reducing
endogenous HDAC6 levels restores learning and memory and α-tubulin
acetylation [183]; (iii) SIRT1 is decreased in the parietal cortex of AD
patients, and the accumulation of Aβ and tau in AD brains might be
related to the loss of SIRT1 [184], since SIRT1 may reduce Aβ production,
activating the transcription of ADAM10 [185]; (iv) in the brains of twins
discordant for AD, trimethylation of H3K9, a marker of gene silencing,
and condensation of heterochromatin structure, are increased in the
temporal cortex and hippocampus of the AD twin as compared to the
twin devoid of AD neuropathology [186]; (v) phosphorylation of H3S10,
a key regulator in chromatin compaction during cell division, is increased
in the cytoplasm of hippocampal neurons in AD cases [187]; (vi) evidence
of DNA damage, as reflected by phosphorylated H2AX at Ser139, is
present in hippocampal astrocytes of AD patients [188]; (vii) LongTerm
Potentiation (LTP) and memory deficits in APP/PS1 transgenic
mice might be mediated in part by decreased H4 acetylation; improving
histone acetylation level restores learning after synaptic dysfunction
[189]; (viii) acetylation of H3 and H4 is increased in 3xTg-AD neurons
relative to non-transgenic neurons [190]; (ix) nuclear translocation of
EP300 interacting inhibitor of differentiation 1 (EID1), a CBP/p300
inhibitory protein, is increased in the cortical neurons of AD patients,
and overexpression of EID1 is reported to reduce hippocampal LTP and
to impair cognitive function via inhibiting CBP/p300 acetyltrasferase
activity and disrupting neuronal structure [191]; (x) memory formation
leads to a transient increase in acetylation on lysine residues within H2B,
H3, H4 [192,193]; (xi) inhibition of HDAC induces dendritic sprouting,
increases synaptic number, and improves long-term memory [194]; (xii)
overexpression of neuronal HDAC2 decreases dendritic spine density,
synapse number, synaptic plasticity and memory formation, and HDAC2
deficiency increases synapse number and memory facilitation [195,196];
(xiii) HDAC4 is involved in learning and synaptic plasticity, and selective
inhibition of HDAC4 activity may deteriorate learning and memory [197];
(xiv) treatment of hippocampal neurons with HDAC inhibitors facilitates
Bdnf expression via hyperacetylation of histones at the Bdnf promoters
[198,199]; (xv) histone(H3K4) methylation participates in the regulation
of Bdnf expression and memory formation [200]; (xvi) histone methylation
also facilitates memory consolidation coupled with histone acetylation;
inhibition of HDACs with Sodium Butyrate (NaB) causes an increase
in H3K4 trimethylation and a decrease in H3K9 dimethylation in the
hippocampus after fear conditioning [200]; (xvii) histone H3 acetylation,
methylation and phosphorylation is increased in the prefrontal cortex of
Tg2576 mice, and histone H4 acetylation is increased in the hippocampal
CA1 neurons of these transgenic mice [2,4,201].
Several lncRNAs are dysregulated in AD (Sox2OT, 1810014B01Rik,
BC200, BACE1-AS, NAT-Rad18, 17A, GDNFOS), Parkinson’s disease
(naPINK1, Sox2OT, 1810014B01Rik, BC200), and Huntington’s disease
(HAR1F, HTTAS, DGCR5, NEAT1, TUG1) [202]. miRNAs belong to
the class of non-coding regulatory RNA molecules of ~22 nt length and
are now recognized to regulate ~60% of all known genes through posttranscriptional
gene silencing (RNA interference) (RNAi). Alterations
in epigenetically-regulated miRNAs may contribute to the abnormal
expression of pathogenic genes in AD [170,202]. Examples of miRNAs
directly linked to AD pathogenesis include miR-34a (1p36.22), miR-34b/c
(11q23.1), miR-107 (10q23.31), miR-124 (8p23.1/8p12.3/20q13.33),
miR-125b (11q24.1/21q21.1), and miR-137 (1p21.3); and examples of
epigenetically regulated miRNAs with targets linked to AD pathogenesis
are let-7b (22q13.1), miR-9 (1q22/5q14.3/15q26.1), miR-132/212
(17p13.3), miR-146a (5q34), miR-148a (7p15.2), miR-184 (15q25.1),
and miR-200 (miR-200b/200a/429, 1p36.33; miR-200c/141, 12p13.31)
[2,4,170].
Pharmacoepigenetics
Epigenetic regulation is responsible for the tissue-specific expression of
genes involved in pharmacogenetic processes, and epigenetics plays a key
role in the development of drug resistance. In this regard, to optimize
therapeutics with this category of drugs, it is important to understand the
reciprocal effects that epigenetic drugs exert on pathogenic, mechanistic,
metabolic, and transporter genes [2,3,86]. Although this is a still poorly
explored field, epigenetic regulation of genes encoding drug-metabolizing
enzymes (CYP1A1, 1A2, 1B1, 1A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19,
2D6, 2E1, 2J2, 2F1, 2R1, 2S1, 2W1, 3A4, 3A5, 3A7, 3A43, UGT1, GSTP1),
drug transporters (ABCB1/MDR1/P-gp, ABCC1/MRP1, ABCC11/MRP8,
ABCG2/BCRP, SLC19A1, SLC22A8), and nuclear receptors (RARB2,
ESR1, NR1I2, HNF41) has been documented in pioneering studies of
pharmacoepigenetics [86,203,204].
Conclusions
- AD is a polygenic/complex disorder in which multiple genomic
defects, epigenetic changes, and environmental factors are
potentially involved.
- The interplay of pathogenic, mechanistic, metabolic, transporter, and
pleiotropic genes is responsible for the therapeutic response in AD.
- Epigenetic phenomena (DNA methylation, histone modifications,
chromatin remodeling, and miRNA dysregulation) may also affect
the pharmacogenetic outcome.
- Different APOE-associated haplotypes influence the pharmacological
effect (efficacy, safety) of drugs in AD.
- Only 25% of the Caucasian population are extensive metabolizers
for drugs metabolized via CYP2D6-CYP2C9-CYP2C19 enzymes.
- CYP2D6 poor (PM) and Ultra-Rapid Metabolizers (UM) are the
worst responders to drugs in AD; and there is a tendency for the
accumulation of PMs and UMs among patients harboring the
APOE-4 allele.
- APOE-4 carriers are the worst responders and APOE-3 carriers are
the best responders to conventional treatments.
- TOMM40 poly T-S/S carriers are the best responders, VL/VL and
S/VL carriers are intermediate responders, and L/L carriers are the
worst responders to treatment.
- Patients harboring a large (L) number of poly T repeats in intron 6 of
the TOMM40 gene (L/L or S/L genotypes) in haplotypes associated
with APOE-4 are the worst responders to treatment.
Patients with short (S) TOMM40 poly T variants (S/S genotype), and to
a lesser extent S/VL and VL/VL carriers, in haplotypes with APOE-3 are
the best responders to treatment. In 100% of the cases, the L/L genotype
is exclusively associated with the APOE-4/4 genotype, and this haplotype
(4/4-L/L) is probably responsible for early onset of the disease, a faster
cognitive decline, and a poor response to different treatments.
