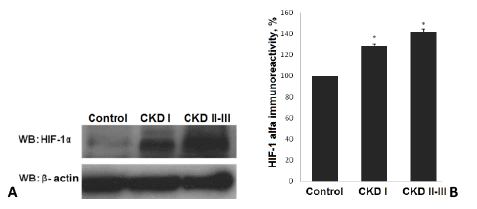
Figure 1: Level of HIF-1α in plasma of patients with nephritic syndrome and control. A - Level HIF-1α; B -HIF-1α immunoreactivity; * - p<0,05. WB – Western Blotting.

Ievgeniia Burlaka*
National O.O. Bogomolets Medical University, Department of Pediatrics № 4, Kyiv, Tolstogo street 10, 01004, Ukraine*Corresponding author: Ievgeniia Burlaka, MD, PhD. National O.O. BogomoletsMedical University, Department of Pediatrics №4, Kyiv, Tolstogo street 10, 01004, Ukraine, Tel: +38- 097-432-49-44; E-mail: evgbur1982@gmail.com
Increased albumin concentration in primary urine leads to apoptosis and fibrosis of podocytes and tubule cells and is a main cause of functional deterioration in chronic kidney disease. Here we show that excessive albumin uptake into rat primary renal cells causes an almost immediate mitochondrial accumulation of the apoptotic factor Bax.
We show that nephrotic patients reveal a high level of marker of cellular hypoxia HIF-1αand its dependence on the level of kidney function impairment and proteinuria. The progression of the sclerosis as a sign of irreversible kidney damage is accompanied by gradual increase in expression of proapoptotic factor Bax.
We conclude that albumin-induced disorders in nephrotic children are presented by disturbances in apoptosis controlling system and accompanied by cellular hypoxia.
Nephrotic syndrome; Albumin; Toxicity; HIF-1α; Bax; Immunostaining
Albuminuric kidney diseases accompanied by Chronic Kidney Disease (CKD) are a rapidly increasing world-wide public health problem. CKD results from a variety of causes, including diabetes, glomerulonephritis, hypertension, infections and polycystic kidney disease [1]. Most forms of CKD are progressive [1,2] and are characterized by disrupted glomerular perm-selectivity [3], glomerular sclerosis, albuminuria, loss of podocytes and glomerular tubular disconnection [4,5]. Albuminuria, which is a well-documented predictor of the progressive loss of kidney function, is also considered to be a cause of structural kidney damage and loss of function [6-9].
The activation of the intrinsic mitochondrial apoptotic pathway is initiated by the accumulation of Bax, a pro-apoptotic member of the Bcl2 family, in the outer mitochondrial membrane, where it oligomerizes and penetrates the inner mitochondrial membrane [10]. Here we report the time sequence for mitochondrial Bax accumulation, depolarization of the mitochondrial membrane following albumin uptake into rat primary proximal tubule cells (RPTC).
Main pathomorphological outcomes that apply to kidney damage in CKD are glomerulosclerosis, vascular sclerosis, tubule-interstitial fibrosis. Adaptive changes of nephrons after the primary injury whichcan no longer be compensated with time, ultimately lead to irreversible disorders - scarring, sclerosis and further loss of nephrons leading to the end-stage CKD (ES-CKD) formation [5]. Inflammation plays an important role in the development and progression of chronic kidney pathologies and is the primary and persistent violation, which underlies the pathogenesis of others. Renal histology in chronic renal pathologies including nephrotic syndromeis characterized by typical signs of inflammation i.e. infiltration with white blood cells, hyperemia, fibrosis etc. In addition to inflammation, fibrosis has a role in nephrotic syndrome. These disorders are accompanied by activation of the renin-angiotensin-aldosterone system, oxidative stress, endothelial dysfunction and others [1-3]. Allmentioned above pathophysiological violations might beacompanied by apoptosis. Apoptosis is programmed cell death that occurs when kidney disease has a place and plays an important role in their physiology. Harmful effects of apoptosis are in fact a source of a large number of kidney cells lost during and/or renal inflammation, scarring, loss of kidney function [11]. The molecular mechanisms underlying irreversible renal damage in children with nephrotic syndromedepending on apoptosis activation might be a potential therapeutic issue in CKD treatment.
An examination of renal biopsies of 53 patients (aged 10 to 15 years) with nephrotic syndrome hospitalized in Pediatric Nephrology unit of the Children Clinical Hospital №7 (Kyiv, Ukraine) was done. All patients were treated accordingly the local protocols. Among all patients 24 (45,28%) were with hormone-sensitive type of nephrotic syndrome, others 29 (54,72%) showed hormone-dependent type of nephrotic syndrome.Complex examination other than conventional methods (inspection, monitoring blood pressure, general and biochemical blood tests, determination of daily proteinuria, urinary sediment study and concentration ability of the kidneys, ultrasound of the abdomen etc.), immunohistochemical assessment of apoptosisdependent glomerular and tubule-interstitial damage were done. The level of kidney function impairment (stage of Chronic Kidney Disease, CKD) was assessed by the value of glomerular filtration rate (GFR). GFR was calculated by Schwartz formula. Stage I CKD refers to normal or high GFR (GFR >90 mL/min); stage II CKD - GFR = 60-89 mL/min; stage IIICKD - GFR = 30-59 mL/min; stage IV CKD - GFR = 15-29 mL/ min; stage V CKD - GFR <15 mL/min.
Renal tissue (3 μm sections) was deparaffinized and rehydrated prior to processing. Antigen retrieval was carried out by boiling in Citrate Buffer (10 mM Citric Acid, 0.05% Tween 20, pH 6.0) for 20 min. Sections were treated with Triton X-100 0.3% (Sigma-Aldrich NV/SA, Bornem, Belgium) in PBS for 20 min. After three PBS washes, sections were incubated with blocking buffer (5% bovine serum albumin and Triton X-100 0.1% in PBS) for 1 hour. The rabbit polyclonal anti-Bax antibody (Santa Cruz, CA, USA) was applied at a dilution of 1:50 in 5% bovine serum albumin in PBS overnight at 4°C. Following three PBS washes, sections were incubated with a secondary Alexa Fluor 488 goat anti-rabbit IgG (1:500) for 1h at room temperature. Nuclei were counterstained with DAPI. All samples were stained for an identical length of timeand under identical conditions. All recordings for each staining were done during one day using identical gain settings. Sections were mounted in ImmuMount (Thermo Shandon, Midland,Canada) and observed with Leica TCS SP inverted confocal scanning laser microscope using25X/0.8NA oil-immersion objective. Image analysis was performed using ImageJ software (NIH Image, Baltimore, MD, USA). Three areas in each section were analyzed.
Proteins solubilized in Laemmli sample buffer were resolved in polyacrylamide gels by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Membranes were then blocked in 5% non-fat milk in TBS-T (136 mM NaCl, 10 mM Tris, 0.05% Tween 20) and immunoblotted using the Bax and HIF-1α Ab (Cell Signaling Technology, Danvers, MA, USA) and actin mouse mAb (BD, LexingtonKY, USA) for 1 h at room temperature. The actin mouse mAb were used as a loading control. After three washes with TBS-T, the membranes were incubated with secondary anti-rabbit oranti-mouse antibodies labeled with horse radish peroxidase for 1 h at room temperature. Membranes were washed three times with TBS-T. The protein bands were visualized by chemiluminescent substrate ECL. Quantitation of the protein content was done by densitometric analysis.
RPTC were prepared from kidneys of 20-day-old male Sprague-Dawley rats as described previously. The studies performed in Sweden followed the Karolinska Institute regulations concerning care and use of laboratory animals and were approved by the Stockholm North ethical evaluation board for animal research. The kidneys were removed and placed in 0.9% NaCl at room temperature. The cortical layers were dissected and placed in Hank’s balanced salt (Invitrogen, Grand Island, USA) solution at 37°C and gently mixed using a fire-polished Pasteur pipette. The reaction was stopped by washing the cells twice in a solution containing 1% trypsin inhibitor. After washing, equal volumes of cell suspension were plated on 12-mm glass coverslips in 24-well Petri dishes. The cells were cultured for 3 days in supplemented DMEM (20mM HEPES, 24 mM NaHCO3 , 10 µg/ ml penicillin, 10 µg/ml streptomycin, and 10% FBS) on glass coverslips in 5% CO2 at 37°C. On day two in vitro, when the cells have been shown to maintain most of their proximal tubule characteristics, the cells were exposed to the 0, 5, 10, or 20 mg/mL of fatty acid and endotoxin-free bovine albumin alone (Sigma-Aldrich, St.Louis, USA), with ouabain (Sigma-Aldrich, St.Louis, USA) or with vehicle (PBS) for 8 or 18 h.
RPTC were cultured as described previously. On day two in vitro, when they have been shown to maintain most of their proximal tubule characteristics, the cells were exposed to the mitochondria-targeted green fluorescent protein CellLight® Mitochondria-GFP BacMam (Life Technologies, Grand Island, USA) overnight in the incubator. On day three in vitro, RPTC were incubated with 0,2.5 or 10 mg/mL of albumin with or without ouabain (5 nM) or vehicle (PBS) for 0-8 h. In another set of experiments, the cells were treated with 10 mg/mL albumin for 0, 15, 30 or 45 min. For Bax immunostaining, the cells were fixed in 4% PFA, washed once with cold PBS and treated with Triton X-100 (Sigma-Aldrich NV/SA, Bornem, Belgium). The mouse monoclonal anti-Bax [6A7] Ab primary antibodies (Abcam, Cambridge, UK) were applied overnight at 4°C. The controls were subjected to the same treatment, but the primary antibody was omitted. The secondary Alexa Fluor 546 goat anti-mouse IgG IgG (Invitrogen, Grand Island, NY, USA) was applied for 1 h at room temperature. The cells were mounted and observed using a Zeiss LSM 510 laser scanning confocal microscope and a 63X/1.4NA oil objective. Analysis of the Bax translocation to the mitochondria was performed with the Matlab image processing toolbox.
Statistical analysis was done using the method of variation statistics (STATISTICA 6.0) and nonparametric statistical approaches (MannWhitneytest). Results are presented as Mean ± SEM.P<0.05 was considered as statistically significant.
We have detected an increased level of marker of cellular hypoxia (HIF-1 α) in blood serum of all patients with nephrotic syndrome as compared to control group. Moreover, a dependence between the levels of renal insufficiency (assessed by GFR) and cellular hypoxia has been documented. The levels of HIF-1α in group with CKD I st. was detected at level over excided the control group value by 28,6% (P<0.01 compared to control group) and by 41,3% (р<0,01 compared to control group) in patients with CKD II-III st. (Figure 1).
Individual analysis of the HIF-1α levels in serum and correspondent levels of daily proteinuria in patients with nephritic syndrome showed that patients presenting high proteinuria levels also presented high levels of HIF1a (Figure 2).
These data indicate the dependence of chronic hypoxia on the degree of damage to the kidney filtration barrier and is a proof of the direct influence of hypoxia and CKD progression.
We have analyzed the levels of expression and localization of proapoptotic factor Bax in patients with morphological variant of nephrotic syndrome- focal segmental glomerulosclerosis (FSGS). Stages of FSGS were determined by level of glomerular sclerotic area. Level of sclerosis corresponding to ≤ 25% of the glomerular area was assumed as I stage of FSGS, II stage of FSGS - 25-50%, III stage - 50-75% and IV stage- 75-100%. Analysis of Bax expression in kidney biopsies from children with focal segmental glomerulosclerosis shows the presence of high level of Bax expression in both glomerular and tubule-interstitial segments. Higher level of immune signal was recorded in glomeruli as compared to tubule-interstitial segment in FSGS I-II stages (43,57 ± 0,88 a.u. vs 24,9 ± 0,41 a.u., P<0.01). When complete glomerular sclerosis presents a high level of Bax was documented in the surrounding tubule-interstitial segment (13,7 ± 0,42 a.u. vs 22,5 ± 0,65 a.u., P<0.01) (Figure 3).
The intrinsic apoptotic pathway is initiated by the translocation of apoptotic factor Bax to the mitochondria, where it permeabilizes the outer mitochondrial membrane to promote apoptosis [3,11]. In this study we used an albumin to induce apoptosis in RPTC.
Figure 1: Level of HIF-1α in plasma of patients with nephritic syndrome and control. A - Level HIF-1α; B -HIF-1α immunoreactivity; * - p<0,05. WB – Western Blotting.
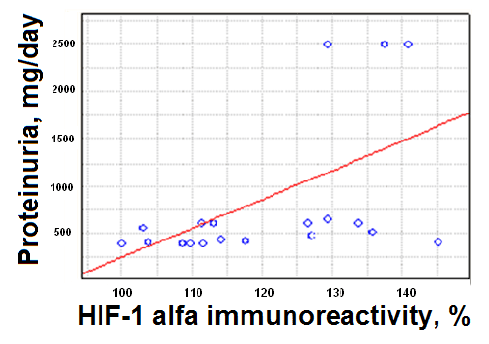
Figure 2: Dependence of HIF-1αlevels on daily proteinuria values in children with nephritic syndrome.
To further characterize mitochondrial involvement in albumin toxicity, we performed time sequence studies, monitoring Bax and mitochondria co-localization in albumin-exposed rat proximal tubule. An albumin was used in concentration 2.5 mg/mL.The co-localization of immunolabeledBax and mitochondria expressing GFP was analyzed in confocal micrographs (Figure 4). Co-localization increased in a time-dependent manner, and the increase was significant after 2 hours incubation with 2.5 mg/mL albumin.
Pathogenesis of the nephrotic syndrome deals with inflammation leading to progression and development of the irreversible tissue damages. Hypoxia is an important link in inflammation development. A mechanism by which HIF may impact the pathogenesis of kidney damage is through the regulation of inflammatory responses. Micro environmental changes, such as hypoxia, strongly impact inflammatory cell recruitment and function [9]. Moreover, hypoxia has been shown to induce apoptosis, where HIF-1 plays a complex role. It has also been demonstrated that the expression of HIF-1α significantly correlated with apoptosis and the proapoptotic factors, such as caspase-3, Fas, and Fas ligand. This finding has been shown on in vitro models [8,9].
The relationship between the two cardinal pro- and anti-apoptotic members of the Bcl2 family of proteins, Bax and Bcl-xL, plays a crucial role in determining the balance between cell life and death. We demonstrated that сellular uptake of albumin causes rapid accumulation of Bax around the mitochondria, resulting in the disabling of the mitochondrial membrane and initiation of the apoptotic process [10].
Here we show that both over expression of HIF-1α and apoptotic factor Bax have a place in children with nephritic syndrome meaning that chronic hypoxia is a factor predisposing disturbances in system controlling apoptosis in this cohort of patients. Our data demonstrate that there is a dependence between the level of hypoxia, Bax overexpression and the stage of CKD. We show that progression of glomerulosclerosis in children with nephrotic syndrome is accompanied by increased activity of proapoptotic factor. Revealed dependence of topology of Bax levels on FSGS degree indicates that development of glomerular and tubule-interstitial disorders under the influence of proteinuria occurs in specific range.
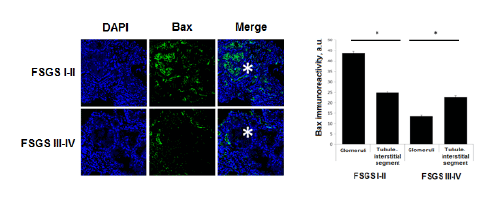
Figure 3: Topical characteristic of the Bax expression in different stages of FSGS.DAPI – visualization of nuclei; Bax – Bax immune signal in kidney tissue. * - glomeruli, * p<0,05.
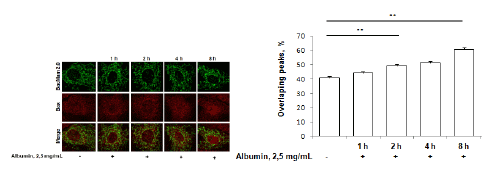
Figure 4: Immunofluorescence staining of proapoptotic factor Bax (red) in RPTC incubated with 2,5 mg/ml albumin. Cells were transfected with mitochondrial marker BacMam 2.0 (green). Number of co-localized Bax/mitochondrial peaks was counted. **p<0.01. Statistical analysis was performed using the Mann-Whitney U test. Experiments were repeated four times.
Further studyof the molecular mechanisms which provides kidney protection in nephrotic patients upon the treatment with agents regulating apoptosis in addition to the conventional therapy has a great importance.
This work has been done with support from nephrology unit of Children Clinical Hospital №7 (Kyiv, Ukraine), this work was covered by the grant 0105U 003769.
Download Provisional PDF Here
Article Type: Research Article
Citation: Burlaka I (2016) Albumin-Induced Primary Disorders in Nephrotic Syndrome: Experimental and Clinical Data. J Mol Med Clin Appl 1(1): doi http://dx.doi.org/10.16966/2575-0305.103
Copyright: © 2016 Burlaka I. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access