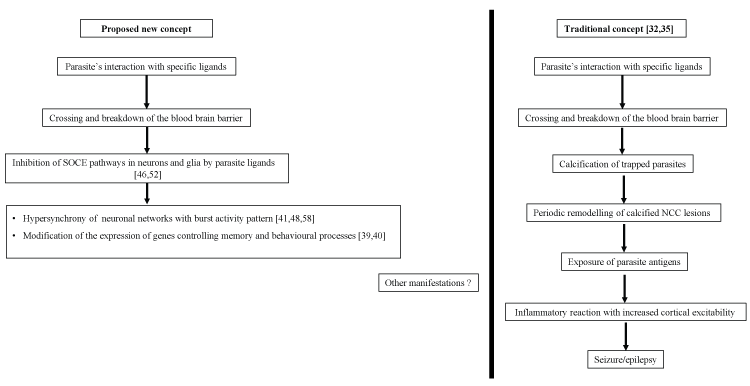
Figure 1: Pathophysiologic concepts (hypothesis) of epilepsy and migraine-like headache in neurocysticercosis (NCC) SOCE: Store-operated Calcium entry; Numbers in brackets stand for corresponding references.
Yannick Fogoum Fogang1* Joseph Kamtchum-Tatuene2 Callixte Kuate-Tegueu3 Paul Chimi Mbonda4 Moustapha Ndiaye1 Amadou Gallo Diop1
1Department of Neurology, Fann teaching hospital, Dakar, Senegal*Corresponding author: Yannick Fogoum Fogang, Department of Neurology, Fann Teaching Hospital, Dakar-Senegal, P.O. BOX: 5035, Tel: +221777390619; E-mail: yanfogang@yahoo.fr
Cysticercosis is the most common helminthic disease of the nervous system in humans. The clinical presentation of neurocysticercosis (NCC) is nonspecific and can mimic a wide array of primary central nervous system (CNS) disorders, making its diagnosis a challenge especially in endemic areas. The pathophysiology of episodic CNS manifestations of NCC is not well understood. We support the hypothesis that mechanisms used by cysticerci to escape the host’s immune system interfere with store-operated calcium entry (SOCE) pathways. This interference may modify brain excitability, leading to episodic manifestations like epilepsy and headaches.
Recent findings suggest that the store-operated calcium entry (SOCE) signaling pathway expressed in host tissues is downregulated by cysticerci ligands. SOCE regulates a vast array of cellular functions in excitable and non-excitable cells including modulation of neuronal excitability and regulation of synaptic plasticity. Inhibition of the SOCE signaling pathway alters synaptic plasticity and synchronization of cortical neuronal networks in vitro and in vivo. These modifications may lower seizure or headache thresholds, increasing the probability of developing these disorders.
This hypothesis could be explored to improve our understanding of the mechanisms involved in episodic manifestations of NCC. If confirmed, potential therapeutic opportunities could be expected from pharmacological modulations of specific proteins in the SOCE signaling pathway
Neurocysticercosis; Epilepsy; Headache; Migraine; Pathophysiology; Store-operated calcium entry; STIM1; Calcium signaling/ homeostasis
CNS: Central Nervous System; CSF: Cerebro-Spinal Fluid; CRAC: Calcium Release-Activated Calcium Channel; ER: Endoplasmic Reticulum; SHE: Syrian Hamster Embryo; LPS: Lipopolysaccharide; LTP: Long Term Potentiation; NCC: Neurocysticercosis; NF- κB: Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells; SOCE: Store-Operated Ca2+ Entry; STIM1: Stromal Interaction Molecule 1; STIM 2: Stromal Interaction Molecule 2; TLR: Toll-Like Receptor.
Cysticercosis is the most common helminthic disease of the nervous system in humans [1]. It is caused by cysticercus cellulosae, the larval stage of the tapeworm Taenia solium. Unlike other infectious agents, helminths are pluricellular organisms which have developed more elaborate mechanisms to escape their host immune system. Helminths have been specifically identified as organisms that can potentially influence both the host immune system and its metabolism [2-4]. They have been a part of the human evolutionary environment for millions of years. Thus, both the current parasites and the human host may have inherited features allowing a complex equilibrium between symbiotic and parasitic mechanisms. Cerebral neurocysticercosis (NCC) is a chronic multistage parasitic infection of the brain. It is highly prevalent in many developing countries, with a prevalence ranging from 2.5% to 6% in the general population in Latin America [5]. During the last few years, the occurrence of NCC has also been increasingly documented in several non-endemic countries due to immigrants or travelers from endemic countries [6]. However, it should be mentioned that the real prevalence of NCC is difficult to establish, given the fact that a significant proportion of patients are asymptomatic [7] and the diagnostic criteria are still under debate [8].
The clinical presentation of NCC can mimic almost all neurological disorders [9]. Seizures and headaches are the main presenting manifestations of NCC [9-11], and it is estimated that NCC accounts for one-third of epilepsy cases in endemic areas [12-18], with a relative risk to develop epilepsy compared to the general population between 2.7- 4.3 [19]. Numerous case reports, case series, and epidemiologic studies have suggested an association between NCC and migraine-like headaches [20-24] though the stronger evidence is still needed to establish a formal causal relationship. Some authors have found a relative risk for patients with NCC to develop recurrent migraine-like headaches between 2.65- 3.39 [24,25], quite similar to that for epilepsy. In another study, persistent or recurrent headaches and seizures following NCC in children were reported in up to one-quarter of patients [10]. It is difficult to distinguish NCC-associated epilepsy or a headache from genetic epilepsies and primary headaches by considering only the clinical presentation and the response to treatment [24,26-28]. In many cases, there is no correlation between seizure semiology, interictal EEG abnormalities, and parasites location [29]. NCC-associated epilepsy is rarely refractory to treatment [26,30] with good prognosis after treatment [29]. Yet, mean age at onset seems higher to that observed in genetic epilepsies, and the frequency of seizures also seems to be higher at onset [23,27,31].
Epidemiological and clinical data suggest that epilepsy or migraine-like headaches associated with NCC may arise from the same phenomenon. However, there is no clear mechanism to explain this. Some authors have proposed that calcified NCC lesions may undergo periodic morphological changes related to a mechanism of neural remodeling. This may expose trapped parasite’s antigenic material to the host immune system, causing inflammatory changes in the brain parenchyma that subsequently lead to seizures, focal neurological deficits, or recurrent episodes of a headache in some patients [24,31-35]. This model is interesting but has some shortcomings. Indeed, it does not explain why there is often no direct topographical relation between the NCC lesion and the epileptic focus. It also does not explain why some patients have seizures or a migraine-like headache without any radiological evidence of an ongoing perilesional inflammatory reaction, especially on brain magnetic resonance imaging studies [29,36]. Consequently, other pathomechanisms should be explored, notably, those involving a modification of neuronal excitability both in cortico-subcortical networks (for epilepsy) and in the trigeminovascular system (for a migraine-like headache). Besides the classical ion channels found in the neuronal membrane, other systems and pathways such as the store-operated calcium entry (SOCE) pathways are involved in the regulation of the intracellular ionic equilibrium and are determinants of the neuronal excitability [37-43].
SOCE is a ubiquitous cell signaling pathway that regulates a vast array of cellular functions [44]. SOCE is initiated by the depletion endoplasmic reticulum (ER) Ca2+ stores, which is detected by stromal interaction molecules (STIM) 1 and 2 that activate selective calcium channels, Ca2+ release-activated Ca2+ (CRAC) channels and transient receptor potential canonical (TRPC) channels [44,45]. Activation of the CRAC and TRPC channels results in a secondary influx of extracellular Ca2+ with a more substantial and sustained increase in cytosolic Ca2+ levels. STIM proteins are expressed in excitable and non-excitable cells [46-48]. They are present in the brain with STIM1 being predominantly expressed in astrocytes and STIM2 in neurons [48].
Herein, we present the hypothesis that mechanisms used by cysticerci to escape the host’s immune system may interfere with store-operated calcium entry (SOCE) pathway. This interference may modify brain excitability, leading to episodic manifestations like epilepsy and headaches (Figure 1).
Figure 1: Pathophysiologic concepts (hypothesis) of epilepsy and migraine-like headache in neurocysticercosis (NCC) SOCE: Store-operated Calcium entry; Numbers in brackets stand for corresponding references.
Helminths can be remarkably efficient at establishing chronic infections although many cases remain asymptomatic [49,50]. Helminthinduced modulation of host’s inflammatory reaction is essential for the parasite to escape the immune response and establish a long-standing infection [12,39]. Meanwhile, the down-regulation of host’s inflammatory reaction is beneficial to host survival as it prevents tissue damage related to inflammation. In NCC, the immunosuppressive effects induced by viable cysticerci contribute to a long asymptomatic phase [14,50,51]. The immune modulatory effects of helminths are marked, and both parasitespecific antigens and different levels of immune suppression are well documented in human studies [52]. During acute infection, antigenspecific T-cell responses are initially stimulated and cells proliferate in response to parasite antigens. With increasing exposure of the immune system to parasite antigens, the immune system becomes increasingly hyporesponsive, first to parasite-specific antigens and subsequently to bystander antigens when high worm burden occurs. Curative chemotherapy restores antigen-specific responses [52].
The mechanism of immunosuppression in NCC has been recently studied. Induction of the inflammatory response to various stimuli has been shown to require increased cytoplasmic Ca2+ turnover for proper signal transduction [53]. At the cellular level, the onset of Ca2+ signaling is marked by an increase in cytosolic Ca2+ through the release of Ca2+ from intracellular endoplasmic reticulum (ER) stores as well as influx across the plasma membrane [53]. This increase in intracellular Ca2+ triggers activation of downstream signaling pathways leading to inflammatory response [54]. In a study using a murine model for NCC [46,55-57], a defect in microglia and myeloid cell activation/maturation in helminthinfected brains was observed. Moreover, cestode’s soluble antigens inhibited Toll-like receptor (TLR)-ligand-induced pro-inflammatory cytokine production and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation in vitro. Additionally, exposure to parasite ligand also inhibited non-TLR agonist induced (thapsigargin exposure) activation of Ca2+ signaling pathway [46]. Helminth antigens control host immune response by inhibiting cell Ca2+ entry through store-operated calcium entry (SOCE) signaling pathways [46,47]. SOCE signaling pathway is ubiquitous and is also present in many other tissues like the nervous system. Thus, SOCE pathway dysfunction may lead to collateral effects on immune and non-immune CNS cells (neurons, glial cells) and cause brain dysfunction.
Inhibiting SOCE with Lanthanum attenuates spontaneous Ca2+ transients at the synaptic level. They are important for short-term synaptic plasticity and may also contribute to long-term plasticity [41]. Inhibition of SOCE with 2-aminoethoxydiphenyl borate (2-APB) and SKF-96365 in hippocampal preparations accelerates the decay of NMDA-induced Ca2+ transients without affecting their peak amplitude. This inhibition also attenuates tetanus-induced dendritic Ca2+ accumulation and LongTerm Potentiation at Schaffer collateral-CA1 synapses [58], suggesting a link between SOCE and neuroplasticity. SOCE inhibition synchronizes network activity of cortical neurons in culture [48]. Furthermore, inhibition of SOCE promotes burst activity in epileptic hippocampal slice cultures [48], and increases neuronal burst firing in dorsal root ganglion [42].
Genetic and epigenetic changes can be observed in helminth-infected tissues [43]. In Taenia solium infections, that can be associated with brain and hematological malignancies, increased frequency of DNA damage in peripheral blood lymphocytes has been observed [43,59]. Cysticerci may cause host genome damage via other non-inflammatory mechanisms. RNA-mediated damage of DNA in T. solium infection has been described [43] and are known to release an RNA factor that could transform Syrian hamster embryo (SHE) fibroblasts in vitro [60,61]. Whether some of these effects are mediated via an SOCE-dependent pathway is to be determined.
Calcium channelopathies have been largely reported in CNS disorders including epilepsy, migraine, and behavioral disorders amongst others [62]. However, data on cell calcium homeostasis perturbations through SOCE pathways dysfunction are scarce. The available evidence could help to depict the relationship between episodic or transient CNS disorders and neurocysticercosis, consistently found in epidemiological studies.
Stormorken syndrome is a rare autosomal dominant disease first reported in 1985 [63]. Patients present with the mild bleeding tendency, thrombocytopathy, mild anemia, asplenia, tubular aggregate myopathy, myosis, ichthyosis and migraine-like headaches. The STIM1 mutation found in patients with Stormorken syndrome is located in the coiledcoil 1 domain which might serve to keep STIM1 inactive. In agreement with a possible gain-of-function mutation in STIM1, resting Ca2+ levels are elevated in platelets from the patients compared with controls, and SOCE signaling is markedly attenuated [64]. SOCE signaling attenuation can be attributed to high cytosolic Ca2+ levels that may reduce the gradient of Ca2+ concentration across the cell membrane, inhibiting further Ca2+ entry. In other words, STIM1 mutation found in Stormorken syndrome has the same functional consequences with changes induced by NCC on neural and immune cells. This might be a clue that migraine-like headaches observed in patients with NCC are related to an alteration of the SOCE pathway.
A migraine and epilepsy are complex and heterogeneous disorders, in which genetic and environmental factors interact to generate dysfunctions at various levels of the central nervous system. These disorders have a genetic polymorphism which determines a dynamic threshold that can be modified by non-genetic factors such as psychological stress, sleep deprivation, neuroinflammation, hormonal changes, hypoglycemia, and drugs. Altered cortical excitability is a key feature in the pathophysiology of epilepsy. In a migraine also, impaired cortical excitability has been established using clinical neurophysiology methods [65]. SOCE pathway dysfunction induced by NCC in neural networks modifies Ca2+ signaling and cortical excitability [48,66]. Although the mechanisms through which these modifications lead to epilepsy and episodic migraine-like headaches are not well understood, altered cortical excitability leading to reduced seizure or headache threshold may play a key role. Increased synchronization of neuronal networks found with altered SOCE signaling pathways [48] may also trigger thalamocortical dysrhythmia, as seen in conditions such as central cortical neurogenic pain, epilepsy and neuropsychiatric conditions [67]. Many authors suggest that thalamocortical dysrhythmia may also account for dysfunction of cortical sensory information processing seen in a migraine [67,68].
Despite the low inflammatory response, neurocysticercosis has been associated with recurrent seizures and headaches often difficult to distinguish from corresponding primary central nervous system disorders. Symbiotic mechanisms developed by parasites throughout evolution are responsible for collateral and prolonged dysfunctions in host tissues. In cerebral tissue especially, chronic dysfunction in Ca2+ signaling through SOCE pathway may modify neuronal networks excitability, with an increased susceptibility to developing primary-like central nervous system events. Our hypothesis may be complementary to other existing models trying to elucidate the complex pathophysiology of episodic manifestations of NCC (Figure 1). Further research is needed to clarify key issues and should focus on:
With the growing body of research data in biomedical sciences, new approaches are needed to solve complex problems and a systems biology approach where multiple levels of information are integrated is becoming more important in complex disease modelling.
The authors declare that they have no competing interests.
Download Provisional PDF Here
Article Type: Review Article
Citation: Fogang YF, Kamtchum-Tatuene J, KuateTegueu C, Mbonda PC, Ndiaye M, et al. (2017) The Potential Role of Store-Operated Calcium Entry (SOCE) Pathways in the Pathophysiology of Epilepsy and Migraine-Like Headaches in Patients with Neurocysticercosis. J Neurol Neurobiol 3(1): doi http://dx.doi.org/10.16966/2379-7150.137
Copyright: © 2017 Fogang YF, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access