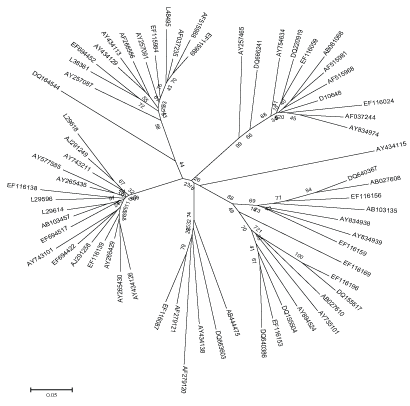
Figure 1: Neighbor-joining tree based on HCV NS5B region.
Amjesh R1,2* Achuthsankar S Nair1 Sugunan VS1
1Department of Computational Biology and Bioinformatics University of Kerala, Thiruvananthapuram, India*Corresponding author: Amjesh R, Department of Computational Biology and Bioinformatics University of Kerala, Thiruvananthapuram, India, E-mail: amjesh@gmail.com
Hepatitis C Virus (HCV) infection is a major health problem that leads to cirrhosis and hepatocellular carcinoma. World over, more than 270-300 million people are estimated to be infected with the virus. HCV is a positive sense single stranded RNA virus and replicates within the cytoplasm of the hepatocyte using its own RNA dependent RNA polymerase (RdRp). RdRp does not have proof reading capacity, and hence generates mutants of the virus, resulting in a chronic infection, which ultimately ends in hepatocellular carcinoma. Such mutations have given rise to several genotypes, subtypes, strains and variants with significant difference in disease outcomes. The mutation rate varies among genotypes, subtypes, strains or even in different sites of the genome. Yet, the extent of heterogeneity is usually moderate, so that estimates of the time of divergence can be computed. The evolution of variants seems to be influenced by the genetic make-up and the immune response of the host and has geographical significance. Here we used phylogenetic analysis and Computational molecular dating techniques to conclude that the ancestral genotype is 7a and that it originated in Canada 363 years ago. Molecular dating was based on the fact that the rate of mutation across all evolutionary lineages is constant over time. Surprisingly, our analyses show that genotype 1d isolated from Canada 5 is the most recent with an evolutionary date of just 33 years. It is evident that HCV is still an emerging virus and demographical parameters seem to have a very strong influence in its evolution. We believe that this emphasizes the need for developing drugs that are customized to act against strains that evolve and become geographically endemic.
Hepatitis C virus; RNA dependent RNA polymerase; Molecular evolution; Evolutionary distance.
Even though the Hepatitis C virus was discovered 25 years ago, its origin remained ambiguous as no closely related viruses have been identified. It infects only humans and in experimental conditions the chimpanzees too. Understanding the history of its evolution would give insight into pathogenicity and predicting its future evolutionary trend would help in formulating strategies to manage the newly emerging strains of the virus. It is very important to understand the origin and evolution of the virus as it has considerable medical significance not just for this disease, but also for other viral diseases. A chronology of the evolution through computed molecular dating techniques would also help in tracing the origin of the virus. Knowledge of viral diversity will help in determining the proper treatment regime for the long-term chronic infection as well as for developing successful anti-viral drugs. Molecular dating approach can also be extrapolated to forecast the evolution of newer strains of the virus.
A comparable hypothesis is the case of HIV which is suggested to have been transmitted to humans from Rhesus monkeys [1]. Tribal Africans who live in close association with these monkeys and who also consume them as raw or un-cooked foods are thought to be the first to get infected with HIV. Extrapolating this observation, many of the viruses which attack humans are considered to have been transmitted from closely associated animals or other lower organisms. The recent transinfection by emerging viruses across different classes of organisms to humans such as the avian influenza virus, swine flu, monkey fever etc. are classic examples that strengthen these observations. Even though it is possible that a cross-species transmission might have occurred from chimpanzees to humans supported by the fact that it has the ability to infect chimpanzees (experimentally proved) no such incidence or clue of natural transmission has been reported or proved.
However Kapoor et al. [2] reported that a single stranded RNA virus which belongs to genus Hepacivirus infects the very close friend of humans “the dogs” and causes pulmonary infection in dogs. These viruses are called Canine hepacivirus (CHV) shares homologous sequences with HCV. This information paved a new approach for understanding the ancestry of HCV. The whole genome of CHV has also been sequenced by Kapoor et.al. The discovery of CHV and its homology with HCV was interesting enough to prompt the search for the existence of related genotypes of HCV which would link it with CHV or any other ancestral viruses. This was done through a molecular dating study of the different genotypes and subtypes that are available on the databases. The main objective of this work was to identify the ancestral genotype of HCV by back-tracking the predecessors of the present HCV genotype from all currently available sequences.
Determining the genotype of HCV is essential for proper disease management. It also helps in monitoring of epidemiological trends and biological features of the virus. Whole genome sequencing and post sequencing analysis are required for identifying the genotype and subtypes of the virus. Nucleotide sequence of certain conserved regions like core, envelope and NS5B have also been used to genotype HCV [3]. Evolutionary relationships were traced with the nucleotide sequence of these regions too.
In order to find the ancestral genotype of HCV, NS5B region of all the available genotypes were selected. NS5B gene sequences were collected from HCV sequence database using the sequence search interface operated by Los Alamos National Security, U.S. Department of Energy’s National Nuclear Security Administration [4]. 65 sequences were selected (single sequence from each available subtypes), and downloaded in FASTA format from Genbank. The sampling date, sampling country and gene identification numbers of these genes are shown in Table 1. The whole genome of CHV were retrieved from Genbank (Accession code: JF744991) for tracking the evolutionary relationship with the HCV.
| Sl. No | Geographical location of sample | Sampling Date | Accession No. | Gene Index No. | Genotype |
| 1 | Berlin | 2001 | AF037244 | gi|3170059 | 2d |
| 2 | Cameroon | 1995 | L38361 | gi|1066643 | 1e |
| 3 | Cameroon | 1998 | AY257087 | gi|30525610 | 1h |
| 4 | Cameroon | 1998 | AY257091 | gi|30525618 | 1l |
| 5 | Cameroon | 2003 | AY265435 | gi|30385487 | 4e |
| 6 | Cameroon | 1995 | L29596 | gi|476675 | 4f |
| 7 | Cameroon | 2004 | AY743211 | gi|54632752 | 4k |
| 8 | Cameroon | 1998 | AY265429 | gi|30385475 | 4p |
| 9 | Cameroon | 1998 | AY265430 | gi|30385477 | 4t |
| 10 | Canada | 2007 | EF115984 | gi|134038120 | 1c |
| 11 | Canada | 2007 | EF115989 | gi|134038130 | 1d |
| 12 | Canada | 2007 | AY434129 | gi|38147572 | 1j |
| 13 | Canada | 2007 | AY434113 | gi|38147545 | 1k |
| 14 | Canada | 2007 | EF116024 | gi|134038200 | 2e |
| 15 | Canada | 2007 | AY754634 | gi|54610706 | 2m |
| 16 | Canada | 2007 | EF116059 | gi|134038270 | 2r |
| 17 | Canada | 2000 | AF279121 | gi|9230780 | 3b |
| 18 | Canada | 2007 | EF116087 | gi|134038326 | 3g |
| 19 | Canada | 2000 | AF279120 | gi|9230778 | 3h |
| 20 | Canada | 2007 | AY434138 | gi|38147587 | 3i |
| 21 | Canada | 2007 | EF116138 | gi|134038428 | 4b |
| 22 | Canada | 2007 | EF116139 | gi|134038430 | 4l |
| 23 | Canada | 2007 | AY434126 | gi|38147567 | 4q |
| 24 | Canada | 2007 | EF116196 | gi|134038544 | 6e |
| 25 | Canada | 2007 | EF116156 | gi|134038464 | 6h |
| 26 | Canada | 2007 | EF116159 | gi|134038470 | 6l |
| 27 | Canada | 2007 | AY894524 | gi|60477635 | 6o |
| 28 | Canada | 2007 | EF116153 | gi|134038458 | 6r |
| 29 | Canada | 2007 | EF116169 | gi|134038490 | 6s |
| 30 | Canada | 2007 | AY434115 | gi|38147548 | 7a |
| 31 | China | 2002 | AY834974 | gi|56123633 | 2f |
| 32 | China | 2002 | AY834938 | gi|56123561 | 6k |
| 33 | China | 2002 | AY834939 | gi|56123563 | 6n |
| 34 | Egypt | 2002 | EF694452 | gi|158146862 | 1g |
| 35 | Egypt | 1999 | AB103457 | gi|40714114 | 4a |
| 36 | Egypt | 2002 | EF694517 | gi|158146992 | 4m |
| 37 | Egypt | 2002 | EF694422 | gi|158146805 | 4o |
| 38 | France | 1999 | AF515988 | gi|29365804 | 1b |
| 39 | France | 1996 | L48495 | gi|1237395 | 1i |
| 40 | France | 1999 | AF515981 | gi|29365790 | 2c |
| 41 | France | 1997 | AF515968 | gi|29365764 | 2i |
| 42 | France | 2006 | DQ220919 | gi|82704304 | 2j |
| 43 | France | 2005 | AJ291258 | gi|11322297 | 4d |
| 44 | France | 2005 | AJ291249 | gi|11322279 | 4h |
| 45 | France | 2004 | AY743101 | gi|54632532 | 4n |
| 46 | Gabon | 1995 | L29614 | gi|476686 | 4c |
| 47 | Gabon | 1995 | L29618 | gi|476688 | 4g |
| 48 | Guinea | 2001 | AF037235 | gi|3170041 | 1m |
| 49 | Japan | 2008 | D10648 | gi|221674 | 2a |
| 50 | Laos | 2004 | AY735101 | gi|52547281 | 6q |
| 51 | Martinique | 2004 | AY257465 | gi|30720399 | 2l |
| 52 | Myanmar | 2007 | AB103135 | gi|47826476 | 6m |
| 53 | Pakistan | 2009 | AB444475 | gi|225380383 | 3k |
| 54 | South Africa | 2001 | DQ164544 | gi|76576168 | 5a |
| 55 | Taiwan | 1993 | DQ666241 | gi|110430931 | 2b |
| 56 | Taiwan | 2005 | DQ663603 | gi|111082412 | 3a |
| 57 | Thailand | 1999 | AB027610 | gi|6136892 | 6c |
| 58 | Thailand | 2006 | DQ640386 | gi|109676985 | 6f |
| 59 | Thailand | 2006 | DQ640367 | gi|109676947 | 6i |
| 60 | Thailand | 1999 | AB027608 | gi|6136888 | 6j |
| 61 | Uganda | 2006 | AY577585 | gi|48995479 | 4r |
| 62 | US | 1984 | AF268586 | gi|13344980 | 1a |
| 63 | Uzbekistan | 2002 | AB081066 | gi|22122154 | 2k |
| 64 | Vietnam | 2006 | DQ155517 | gi|73765290 | 6d |
| 65 | Vietnam | 2006 | DQ155504 | gi|73765264 | 6p |
Table 1: Details of HCV NS5B regions included in this study.
The evolutionary distances were arrived at by tracking the number of changes between nucleotide sequences sampled at different times [5, 6]. Pair wise distance measurement gave an estimate of the evolutionary distance in terms of number of nucleotide substitutions.
The genetic distance was calculated based on Kimura 2 parameter [7] implemented in MEGA software [8]. This was done by estimating transition and transversion differences in nucleotide sequences. The transition type tries to get the difference between both purines and pyrimidines (T↔C, A↔G). In the latter case it computes the distance between one of the two in which one is a purine and the other one is a pyrimidine (T↔A, T↔G, C↔A, and C↔G). The method of calculation is defined in the equation given below.
\[K = - \frac{1}{2}lo{g_e}\{ \left( {1 - {\rm{ }}2P - Q{\rm{ }}} \right)\sqrt {1 - 2Q\} } \]
The fractions of the nucleotide site of transition and transversion were represented by P and Q of the two sequences..
Phylogenetic analysis was used to estimate the evolutionary relationships among groups of organisms or within species. The evolutionary relationship is usually depicted as a tree like diagram know as phylogenetic tree. All the 65 sequences were aligned and converted to PHYLIP format using Clustal W [9]. As the rates of mutations were found to be high in HCV the trees were constructed using DNA Parsimony (Dnapars) program implemented in PHYLIP package [10]. Dnapars assumes that different lineage evolve independently. To confirm the reliability of the phylogenetic tree 1000 bootstrap resampling tests were performed using Seqboot program. It produced a collection of trees rather than a point estimate of an optimal tree. Since such a tree with no measure of reliability is not particularly helpful, a consensus tree was built from out tree file of Dnapars using Consense program. The tree was drawn by the program Drawgram. The ancestral genotype of HCV was then computed by tracing back to a hypothetical genetic sequence from which the evolution of HCV would have commenced.
The hypothetical ancestral sequences of the each node of the phylogenetic tree were estimated by Dnaml program implemented in Phylip. Then the distances from the ancestral sequences to each strain were estimated by the Neighbor-Joining tree and Minimum Evolution tree implemented in MEGA 4. The mean distance was then estimated from distance values obtained from MEGA Neighbor- Joining tree and
Minimum Evolution tree. The molecular date was estimated by a simple division of genetic distance by calibration rate (nucleotide substitution per site per year). The nucleotide substitution rate of HCV was estimated at 0.67*10-3 per site per year [11].
A new sequence data set comprising of all the 65 NS5B region of HCV along with the full genome of CHV was compiled to perform a multiple sequence alignment. Till date only core, NS3 and polyprotein regions of CHV were isolated and sequenced which are made available at Genbank. In the present study, the full genome of HCV was used for the analysis. These alignment files were used to predict the evolutionary distance as mentioned in 2.2.3. A phylogenetic tree was also constructed using a Neighbor-Joining method implemented in MEGA software [12].
The evolutionary distance of HCV were calculated using all the available sequence data of HCV NS5B region as mentioned in Table 2. The evolutionary history inferred using the Neighbor-Joining method and Maximum Evolution tree is shown in Figure 1 and 2. The trees are drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The distances are recorded from these trees of all the strains to their most recent common ancestor. The two methods were adopted to validate the results, as the differences between NJ and ME trees are substantial. The NJ distance and ME distance has slight variations hence the average has been taken to estimate the divergence time. The divergence time calculated using both the mean values of neighbor joining tree as well as maximum evolution tree is shown in Table 2.
Figure 1: Neighbor-joining tree based on HCV NS5B region.
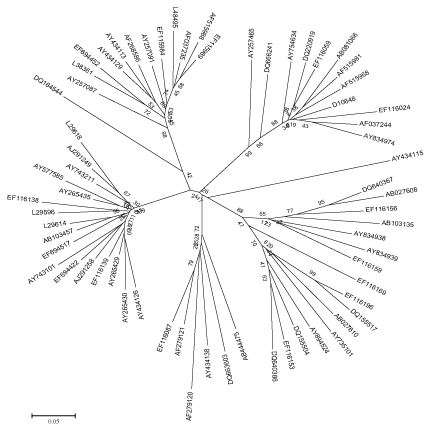
Figure 2: Maximum Evolution tree based on HCV NS5B region.
| Sl. No | Genotype | Accession No. | NJ* Dist. | ME* Dist. | Mean value | Divergence Time |
| 1 | 2d | AF037244 | 0.057 | 0.036 | 0.047 | 70 |
| 2 | 1e | L38361 | 0.068 | 0.086 | 0.077 | 115 |
| 3 | 1h | AY257087 | 0.093 | 0.096 | 0.095 | 142 |
| 4 | 1l | AY257091 | 0.056 | 0.026 | 0.041 | 61 |
| 5 | 4e | AY265435 | 0.031 | 0.026 | 0.028 | 42 |
| 6 | 4f | L29596 | 0.071 | 0.042 | 0.056 | 84 |
| 7 | 4k | AY743211 | 0.044 | 0.021 | 0.032 | 47 |
| 8 | 4p | AY265429 | 0.027 | 0.026 | 0.026 | 39 |
| 9 | 4t | AY265430 | 0.064 | 0.053 | 0.058 | 87 |
| 10 | 1c | EF115984 | 0.061 | 0.031 | 0.046 | 68 |
| 11 | 1d | EF115989 | 0.030 | 0.015 | 0.023 | 33 |
| 12 | 1j | AY434129 | 0.046 | 0.020 | 0.033 | 49 |
| 13 | 1k | AY434113 | 0.054 | 0.026 | 0.040 | 60 |
| 14 | 2e | EF116024 | 0.109 | 0.096 | 0.103 | 153 |
| 15 | 2m | AY754634 | 0.085 | 0.075 | 0.080 | 119 |
| 16 | 2r | EF116059 | 0.086 | 0.057 | 0.072 | 107 |
| 17 | 3b | AF279121 | 0.068 | 0.064 | 0.066 | 98 |
| 18 | 3g | EF116087 | 0.069 | 0.042 | 0.055 | 83 |
| 19 | 3h | AF279120 | 0.174 | 0.132 | 0.153 | 228 |
| 20 | 3i | AY434138 | 0.120 | 0.086 | 0.103 | 154 |
| 21 | 4b | EF116138 | 0.088 | 0.069 | 0.078 | 117 |
| 22 | 4l | EF116139 | 0.039 | 0.021 | 0.030 | 44 |
| 23 | 4q | AY434126 | 0.062 | 0.052 | 0.057 | 86 |
| 24 | 6e | EF116196 | 0.040 | 0.031 | 0.035 | 53 |
| 25 | 6h | EF116156 | 0.081 | 0.058 | 0.069 | 103 |
| 26 | 6l | EF116159 | 0.101 | 0.053 | 0.077 | 115 |
| 27 | 6o | AY894524 | 0.098 | 0.098 | 0.098 | 146 |
| 28 | 6r | EF116153 | 0.072 | 0.047 | 0.059 | 89 |
| 29 | 6s | EF116169 | 0.014 | 0.097 | 0.056 | 83 |
| 30 | 7a | AY434115 | 0.217 | 0.270 | 0.243 | 363 |
| 31 | 2f | AY834974 | 0.059 | 0.047 | 0.053 | 79 |
| 32 | 6k | AY834938 | 0.096 | 0.080 | 0.088 | 131 |
| 33 | 6n | AY834939 | 0.117 | 0.074 | 0.096 | 143 |
| 34 | 1g | EF694452 | 0.069 | 0.047 | 0.058 | 86 |
| 35 | 4a | AB103457 | 0.043 | 0.036 | 0.040 | 59 |
| 36 | 4m | EF694517 | 0.067 | 0.059 | 0.063 | 94 |
| 37 | 4o | EF694422 | 0.063 | 0.052 | 0.058 | 86 |
| 38 | 1b | AF515988 | 0.050 | 0.047 | 0.048 | 72 |
| 39 | 1i | L48495 | 0.072 | 0.042 | 0.057 | 85 |
| 40 | 2c | AF515981 | 0.055 | 0.058 | 0.056 | 84 |
| 41 | 2i | AF515968 | 0.062 | 0.047 | 0.054 | 81 |
| 42 | 2j | DQ220919 | 0.066 | 0.031 | 0.048 | 72 |
| 43 | 4d | AJ291258 | 0.052 | 0.063 | 0.058 | 86 |
| 44 | 4h | AJ291249 | 0.043 | 0.036 | 0.040 | 59 |
| 45 | 4n | AY743101 | 0.087 | 0.063 | 0.075 | 112 |
| 46 | 4c | L29614 | 0.048 | 0.042 | 0.045 | 67 |
| 47 | 4g | L29618 | 0.076 | 0.086 | 0.081 | 120 |
| 48 | 1m | AF037235 | 0.031 | 0.026 | 0.028 | 42 |
| 49 | 2a | D10648 | 0.051 | 0.015 | 0.033 | 50 |
| 50 | 6q | AY735101 | 0.128 | 0.114 | 0.121 | 181 |
| 51 | 2l | AY257465 | 0.116 | 0.079 | 0.098 | 146 |
| 52 | 6m | AB103135 | 0.111 | 0.042 | 0.076 | 114 |
| 53 | 3k | AB444475 | 0.140 | 0.097 | 0.118 | 177 |
| 54 | 5a | DQ164544 | 0.166 | 0.138 | 0.152 | 227 |
| 55 | 2b | DQ666241 | 0.088 | 0.058 | 0.073 | 108 |
| 56 | 3a | DQ663603 | 0.124 | 0.097 | 0.111 | 165 |
| 57 | 6c | AB027610 | 0.104 | 0.098 | 0.101 | 151 |
| 58 | 6f | DQ640386 | 0.082 | 0.058 | 0.070 | 104 |
| 59 | 6i | DQ640367 | 0.041 | 0.026 | 0.033 | 49 |
| 60 | 6j | AB027608 | 0.064 | 0.052 | 0.058 | 87 |
| 61 | 4r | AY577585 | 0.069 | 0.058 | 0.063 | 95 |
| 62 | 1a | AF268586 | 0.050 | 0.036 | 0.043 | 65 |
| 63 | 2k | AB081066 | 0.076 | 0.081 | 0.078 | 117 |
| 64 | 6d | DQ155517 | 0.056 | 0.036 | 0.046 | 69 |
| 65 | 6p | DQ155504 | 0.082 | 0.042 | 0.062 | 92 |
Table 2: Neighbor-Joining (NJ) distance, Maximum Evolution, Mean distance and the divergence time of HCV.
Based on these data, it was deduced that the genotype 7a (Accession No. AY434115) originated approximately 363 years ago in Canada. Genotype 1d (Accession No. EF115989) seems to be a newly/still emerging strain isolated from Canada, and its evolutionary date was computed as 33years. Another set of phylogenetic analyses were conducted using the same data set along with the full genome of CHV. Surprisingly the result showed that CHV is genetically closer to genotype 7a which was interpreted to be the ancestral genotype of HCV. Not surprisingly, genotype 7a is the prevalent strain in Canada. Figure 3 clearly shows the relation between HCV genotype 7a and CHV.
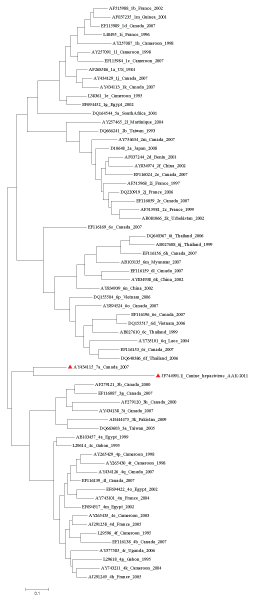
Figure 3: The un rooted Maximum Likelihood tree depicting the phylogenetic relationship among HCV and CHV. HCV genotype 7a and CHV are marked in red.
Despite enormous advances in medical sciences human beings are not able to conquer the bane of viral infections with the help of drugs. Natural immunity alone forms the process by which viral infections are overcome. Any drug or treatment procedure that claims to be effective work by boosting or aiding the immune system to overcome a viral infection.
In the battle between pathogenic viruses and the human immune system an effective strategy enacted by a virus is its constant evolution into a newer species or strains. Such species or strains have been termed “emergent/emerging viral species/strains” and the disease caused is defined as emerging viral disease.
Several newly reported diseases such as bird flu (H5N1), swine flu (H1N1), monkey fever (Kyasanur forest disease) etc. are examples of diseases caused by emerging viruses that have acquired an alarming capability of crossing from one genus to another especially humans.
However at present these example of emerging viruses have not yet succeeded in getting transmitted from one human being to another, or if they do they are weakened to an extent that they do no harm in such trans human infections.
HCV is one of the most dreaded emerging viruses which despite its discovery and description in 1990 has evaded all types of medical interventions till date. Data from this part of the study indicates that HCV too had a trans genus infecting phase from dogs to humans and evolved as an emerging virus approximately 363 years ago, which is an extreme short period in evolutionary time. It is now an emergent virus which has acquired propensity to continuously evolve and thereby defeat all known treatment process as well as the defensive mechanism of the immune system.
Hence it was thought extremely relevant that evolutionary path of HCV should be worked out. In this venture, existing data was put into use which returned the logical and scientific conclusion that the human HCV originated as a trans genus strain (dogs to human) infecting humans approximately 363 year ago [13].
The genotype 7a (Accession no. AY434115) originated approximately 363 years before in dogs in Canada. Genotype 1d (Accession no. EF115989) is the most recently emerged one and their evolutionary date was calibrated as 33 years. In an early report in 1995 J Mellor et al. using a Bayesian analysis proposed that HCV genotypes evolved about 300-400 years ago [14]. The outcome of this study also co relates well with Mellor’s report in a much more scientific and realistic manner.
This study thus proved that the HCV evolved and emerged from CHV, acquired the ability to get transmitted to humans through their best companion ‘dogs’ and latter evolved into a unique viral species that gets transmitted from humans to humans, with the only hurdle that it required a blood-to-blood contact.
Whether it will still evolve and emerge into dangerous strain that over comes this hurdle is a valid but dangerous proposition. This scenario highlights the need for identification of new drugs and treatment procedures that ultimately succeeds in complete eradication of the virus.
The authors greatly acknowledge the members of bioinformatics research team at Department of Computational Biology and Bioinformatics, University of Kerala for fruitful discussions and suggestion which help in completion of this work.
Download Provisional PDF Here
Aritcle Type: Research Article
Citation: Amjesh R (2017) Molecular Evolution Studies on Hepatitis C Virus based on NS5B Region. J Emerg Dis Virol 3(3): doi http://dx.doi. org/10.16966/2473-1846.137
Copyright: © 2017 Amjesh R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access