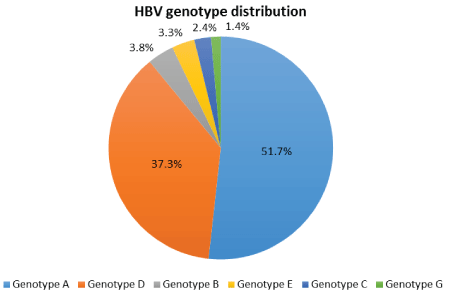
Figure 1: Genotype distribution of South African HBV-positive cohort.
Smuts H1* Sonderup M2 Gogela N2 Spearman CW2
1Division of Medical Virology/NHLS, Department of Pathology, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa*Corresponding author: Smuts H, Division of Medical Virology/NHLS, Department of Pathology, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa, Tel: +27 21 4045306; E-mail: Heidi.Smuts@uct.ac.za
Hepatitis B virus can be divided into 10 genotypes based on sequence heterogeneity. HBV genotype G (HBV-G) has little genetic variation and often co-exists with another HBV genotype. HBV-G infections have been detected around the world, but rarely from Africa. Over a 9 year period, 3 patients were identified with HBV-G infection while attending a specialist Liver Clinic at a tertiary hospital in Cape Town, South Africa. The aim of this study was to describe the clinical and laboratory features of the patients and sequence the full HBV-G genome to determine the phylogenetic relationship with other HBV-G isolates from around the world. Further, co-infection with other HBV genotypes was determined using a multiplex type- specific PCR and cloning. All patients (2 of Caucasian and 1 of Asian ethnicity) belonged to a high risk group of men who have sex with men and were HBeAg positive. Two patients were co-infected with HIV and HBV co-infection (HBV-A) could only be identified in one case. The 2 South African complete genomes, from patients of Caucasian and Asian ethnicity, showed the characteristic features of HBV-G, including the 36 nucleotide insert at the 5’ end of the core gene, 2 stop codons in the precore region and low genetic variability (0.5%). Phylogenetically the genomes clustered together with reference HBV-G strains. This is the first report of the full characterization of HBV-G in South Africa.
Africa; Genotype G; HBV; Clinical; Molecular Characterization
HBV: Hepatitis B Virus; HCC: Hepatocellular Carcinoma; HBeAg: Hepatitis B e Antigen; Pol: Polymerase; HBcAg: Hepatitis B Core Antigen; HBV-G: Hepatitis B Virus Genotype G; BCP: Basal Core Promotor; PC: Precore; MSM: Men who have sex with men.
An estimated 350 million people around the world are chronically infected with hepatitis B virus (HBV) and almost 800000 die from cirrhosis or hepatocellular carcinoma (HCC). The highest prevalence of this vaccine-preventable disease is in sub-Saharan Africa and East Asia where 5-10% of the adult population are chronically infected. In these endemic areas HBV transmission usually occurs in the first 5 years of life from infected mother to infant at birth or horizontally from an infected child to an uninfected one [1].
HBV is a small enveloped partially double-stranded DNA prototype virus of the Hepadnaviridae family. The genome encodes 4 overlapping reading frames: e antigen (HBeAg), core protein (HBcAg), polymerase (Pol) with reverse transcriptase, RNase H and DNA polymerase activity, 3 forms (small, middle and large) of the surface protein and a transcriptional trans activator protein (X) [2].
The lack of proof reading activity of the viral polymerase leads to nucleotide mis-incorporations during replication resulting in sequence heterogeneity. Diversity of >7.5% enables the classification of HBV into 9 genotypes (A-I) [3,4] with a possible 10th (J) based on the sequence of one Japanese patient [5]. Intergroup nucleotide differences of 4-8% allow further classification of genotypes A, B, C, D, F, H and I into 35 sub genotypes [2].
HBV genotypes have a distinct geographical distribution. Genotype A is found in Africa, Europe and the Americas, genotypes B and C predominate in Asia, genotype D has a worldwide distribution, while genotypes F and H occur in South and Central America [4-6]. Sub genotype A1 is found mainly in Africa and accounts for 75% HBV infections in South Africa [7]. Human migrations play a role in the changing geographical prevalence of HBV genotypes and the introduction of new subgenotypes [8,9].
HBV genotype G (HBV-G) was recognized as a new genotype in 2000 [10]. It is distinct from other genotypes in that it has a 36 nucleotide insert at the 5’ end of the core, adding 12 amino acids to the protein. This insertion affects core protein expression, replication and viral secretion [11,12]. In addition, 1 or 2 stop codons at positions 2 and 28 in the precore region prevent the expression of HBeAg [10-13]. Given this, monoinfection with HBV-G is rare as HBV-G replication requires HBeAg to be supplied in trans by co-infection with another HBV genotype such as HBV-C [14], HBV-F [15], HBV-H [16] and often HBV-A [17-19]. A recombinant form may also supply HBeAg [20]. HBV-G is prevalent in certain risk groups such as men who have sex with men (MSM), people who inject drugs and those co-infected with HIV [21,22].
Reports of HBV-G are confined to Europe, the Americas and Japan [16- 18,23,24]. A search of PubMed and GenBank databases for the presence of HBV-G in Africa revealed a single case of this genotype in an HIVinfected person from South Africa in 2005 (accession number EU694179; Mphahlele unpublished). A study of 47 children from Gabon found 2 cases of HBV-G [25] and a patient from Nigeria had the characteristic 36bp HBV-G insertion in the core region in a complex HBV E/D and A3 recombinant virus [26]. These brief reports indicate that HBV-G may be present in Africa. There has also been a suggestion that HBV-G may have a West or Central African origin [27].
This study reports 3 cases of HBV-G detected in a cohort of HBVpositive patients attending the Liver Clinic at a tertiary hospital in Cape Town, South Africa. The complete genomes of the 2 patients were sequenced and compared to HBV-G strains from other regions of the world.
Methods
Over a 9 year period (2007-2015) the genotypes of 209 HBV-positive patients attending the Liver Clinic, Groote Schuur Hospital, Cape Town, South Africa was determined. Three patients (1.4%) were found to be infected with HBV-G (Table 1).
| L-2007 | P-2014 | M-2015 | |
| Age (years) | 32 | 31 | 48 |
| Gender | Male | Male | Male |
| Ethnicity | Caucasian | Asian | Caucasian |
| Risk factor | MSM | MSM | MSM |
| HBV log viral load (IU/ml) 10 |
9,2 | >8,3 | 9,3 |
| HBeAg | Positive | Positive | Positive |
| HBsAg | Positive | Positive | Positive |
| HBV co-infection | ND | HBV-A | None detected |
| HBV serotype | ND | adw2 | adw2 |
| HCV status | ND | ND | Negative |
| HIV status | Positive | Positive | Negative |
| Lymphocyte CD4+ cell count (cells/mm3) | 319 | 347 | ND |
| Liver fibrosis# | Moderate – 3/6 | ND | Severe – 5/6 |
| ALT (U/L) | 202 | 65 | 1043 |
| AST (U/L) | 141 | 66 | 1307 |
Table 1: Demographic, clinical and risk factors of South African HBV-G patients.
Total nucleic acid was extracted from serum/plasma samples using the MagNA Pure LC automated extraction method as per manufacturer’s instructions (Roche Diagnostics GmbH, Penzberg, Germany).
A semi-nested PCR was used to amplify a region of the surface and overlapping polymerase (S/pol) gene: P6F (5’-CCTGCTGGTGGCTCCAGTT-3’) and P2R (5’ – CTAGGAGTTGGCCAGTATGGAT-3’) [28] were used in the first round and P7F (5’-GTGGTGGACTTCTCTCAATTTTC-3’) [29] and P2R used in the second round to yield an outer and nested amplicon size of 1230bp and 1028bp respectively. The PCR was performed on a Swift thermocycler (Esco Micro Pte. Ltd, Singapore) with a 50 µl reaction mixture containing 10 µl extracted DNA, 15 mM Tris-HCL (pH 8), 50 mM KCl, 1.5 mM MgCl2 , 0.2 mM deoxynucleotide triphosphates (ABgene, Epsom, UK), 50 pmol of each primer and 1.5 U Supertherm Taq polymerase (JMR Holdings, Kent, UK). The amplification conditions for the first round were as follows: denaturation at 95°C for 3 minutes, 40 cycles of 95°C for 15sec, 50°C for 25 sec and 72°C for 35 sec followed by a final 72°C elongation step for 7 min. The second round of amplification was the same as above with an increase in the annealing temperature to 55°C. PCR products were visualized by electrophoresis through a 2% agarose gel, ethidium bromide staining and UV illumination.
The whole HBV genome was amplified in 4 overlapping fragments using the universal primers and a modified protocol described by Chook et al. [30]. Four outer and 4 inner primer sets (Sets 1a, 2a, 3 and 4) were used to amplify amplicons of 940bp, 1131bp, 959bp and 748bp respectively which were visualized by electrophoresis through a 2% agarose gel, ethidium bromide staining and UV illumination.
A multiplex type-specific PCR assay [31] was used to detect mixed infections in 2 patients. Primer set N1 targeted HBV genotypes A, B, F and G, while genotypes C, D and E were detected with primer set N2. The presence of more than one band indicated co-infection with 2 or more genotypes.
Cloning of an S/pol PCR product (nt 260-1190) into pGEM-T vector (Promega Corporation, Madison, WI, USA) was performed as per manufacturer’s instructions as a further attempt to identify a mixed HBV infection in patient M-2015.
The S/pol gene and whole genome amplicons were sequenced directly in both directions using PCR specific primers. The Big Dye terminator cycle sequencing kit was used (Applied Biosystems, Foster City CA, USA). The HBV genotype was determined by BLASTn analysis and confirmed using the web-based geno 2 pheno program (http://www.geno2pheno.org/index. php).The HBV whole genome fragments were assembled in DNABaser v.4 (http://www.dnabaser.com). Maximum likelihood phylogenetic trees were constructed in Mega 6.06 [32] using complete HBV-G genomes from Gen Bank and the 2 assembled South Africa sequences with 1000 bootstrap re-samplings.
Nucleotide and amino acid analysis and diversity was performed using Mega 6.06 and the HBV database (https://hbvdb.ibcp.fr/HBVdb) [33].
Common BCP and PC mutations were identified by alignment of the South African HBV-G genomes with a reference HBV sequence.
Resistance mutations in the polymerase gene to the drugs lamivudine, adefovir, entecavir, tenofovir and telbivudine was assessed using the online geno2pheno program (http://www.geno2pheno.org).
DNA methylation in promoter regions may alter gene expression and play a role in the regulation of HBV. The complete genome of the 2 South African isolates were analyzed for CpG islands using the Meth Primer program, (http://www.urogene.org/methprimer) [34] and results compared to other HBV-G sequences.
The SIMPLOT program and boot scanning analysis was used to determine any possible recombination events.
Serotyping of the 2 South African sequences was determined using the online Mutation Reporter Tool (http://hvdr.bioinf.wits.ac.za/tools) [35]. The translated sequences of the HBsAg were analyzed at 5 amino acid positions (122, 127, 140, 159 and 160) which are known to specify serological subtype [36].
The study design conformed to the 2013 Declaration of Helsinki and was approved by the University of Cape Town Human Research Ethics Committee (reference number 761/2016).
The prevalence of HBV genotypes detected in this cohort of patients attending a specialist Liver Clinic at a tertiary hospital was HBV-A (108/209; 51.7%), HBV-D (78/209; 37.3%), HBV-B (8/209; 3.8%), HBV-E (7/209; 3.3%), HBV-C (5/209; 2.4%), and HBV-G (3/209; 1.4%). No genotypes HBV-F, HBV-H, HBV-I or HBV- J were found (Figure 1).
Figure 1: Genotype distribution of South African HBV-positive cohort.
The clinical and epidemiological features of our patients are shown in Table 1. Two patients were co-infected with HIV with a lymphocyte CD4+ count of 319 and 347 cells/mm3 respectively. All were HBe Ag-positive and by implication must have a functioning pre core gene or are co-infected with another HBV genotype to provide a functional core in trans. The viral loads were similar, ranging from log10 8.3–9.3 IU/ml. The patients showed evidence of liver dysfunction with moderate to high Ishak (HAI scores) and elevated liver enzymes. The travel history of these subjects was unknown. This may have provided some evidence as to whether the virus had been acquired locally or abroad.
The complete genome of HBV-G was successfully amplified in 4 overlapping fragments in 2 of the HBV-G-positive patients. No plasma was available for the amplification of the full HBV-G genome of patient L-2007 HBV-G in this patient was identified by sequencing of a 491 bp region of the pol/S region and BLAST n analysis.
The South African HBV-G complete genomes were 3248 nucleotides (nt) in length and showed the characteristic 36 nt insert at the 5’ end of the core protein between nt 1905 and 1942 and 2 stop codons, TAA (nt 1817 instead of CAA) and TAG (nt 1896 instead of TGG), at codon positions 2 and 28 of the precore region, respectively. There was also a 3nt deletion (CAT) in the pre S at nt 2915. Boot scanning of the complete genomes found no recombination events (data not shown) (Accession numbers KY004110-KY004112).
The genetic variability of the South African isolates were compared to 40 full genome sequences in the GenBank database comprising sequences from around the world. The overall genetic distance was 0.1-1.9% (mean 0.5%). Patients M-2015 and P-2014 showed 4 and 7 nt differences respectively to the consensus HBV-G with 4 nt changes common to both isolates, T2057G, C2070T, T2143C, and G2465A resulting in 3 amino acid changes in the core protein (Table 2). P-2014 had an additional 3 nt changes, A636T, A645C and A1764G, which resulted in amino acid changes to the surface protein and X protein (Table 2). In common with all HBV-G sequences the Kozac sequence (nt 1809-1812) was GCAC.
| P-2014 | M-2015 | |||
| Nucleotide | Amino acid | Nucleotide | Amino acid | Protein |
| A636T | Y161F | Surface | ||
| A645C | E164A | Surface | ||
| A1764G | T/I 131A | X |
||
| T2057G | S53A | T2057G | S53A | Core |
| C2070T | S57F | C2070T | S57S | Core |
| T2143C | T2143C | |||
| G2465A | A189T | G2465A | A189T | Core |
Table 2: Nucleotide and amino acid changes in the genome of HBV-G patients.
Analysis of the common BCP/PC mutation sites which infer HBeAg phenotype found that besides the HBV-G signature 1896A mutation, resulting in the TAG stop codon at position 28, patient P-2014 had 1764G compared to all other HBV-G genomes with 1764A.
No resistance mutations to lamivudine or any of the other nucleos(t) ide reverse transcriptase inhibitor drugs, including adefovir, entecavir, tenofovir and telbivudine, were found in the polymerase gene.
The 2 nt changes in the surface gene encoding HBsAg, A636T and A645C (Y161F and E164A), in patient P-2014 were not associated with vaccine escape mutations. Using the Mutational Reporter Tool both patients were assigned a serotype of adw2 based on amino acid residues at positions 122K, 160K,127P, 159A and 160T. All other reference HBV-G isolates are adw2 as well.
The 2 South Africa HBV-G genomes were mapped for the presence of CpG islands I, II, and III. Methylation within these CpG rich islands may play a role in the regulation of gene expression in HBV. Only CpG island II and III were present in both patients spanning nt 1163-1628 and 2350-2494 respectively, which is similar to that found in other HBV-G sequences (Figure 2). However, the size and location of these two islands differed slightly from other HBV-G genomes, particularly CpG island II, which, in the South African genomes and 92.5% (37/40) other HBV-G complete sequences, had an 11 nucleotide gap between nucleotides 1345 and 1356. CpG island II overlaps the enhancer I and X gene promoter, while CpG island III spans the start codon of the polymerase gene.
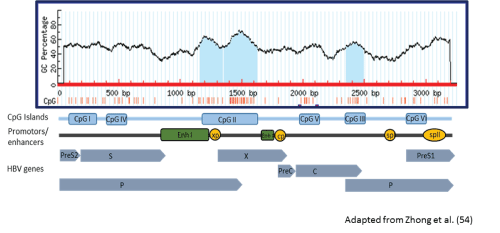
Figure 2: The distribution of CpG islands in a South African HBV-G patient.
The genetic diversity among HBV-G and other genotypes is shown in the maximum likelihood phylogenetic tree (Figures 3A and 3B). The 2 complete sequences of South African HBV-G strains clustered together and with all HBV-G reference sequences available in GenBank (accessed 20 April 2016). The genetic distances, as indicated by branch length, was significantly smaller in HBV-G than found in other genotypes. HBV-G from patient L-2007 formed a distinct branch from the other HBV-G sequences (Figure 3B).
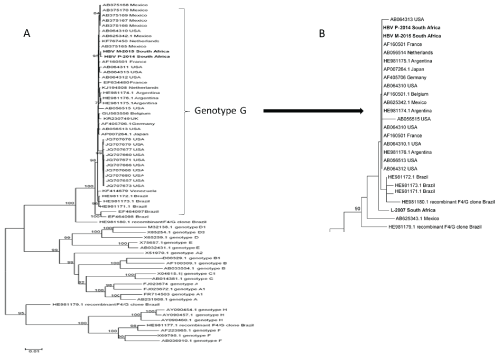
Figure 3: A. Maximum likelihood phylogenetic tree of the full genome of the 2 South African HBV-G genomes and representative sequences of HBV genotypes A-H.
B. Maximum likelihood phylogenetic tree of a 490 fragment of the pol/S region of the 3 South African patients and representative sequences of HBV-G.
To determine if patients M-2015 and P-2014 were co-infected with another HBV genotype, the multiplex genotype-specific method of Liu et al. [31] was employed. PCR products of 508 bp and 228 bp were amplified with the N1 primer set from patient P-2014, indicating that HBV-A was present together with HBV-G. No additional PCR products were detected in patient M-2015. To further investigate whether the latter patient was co-infected, a 930 bp fragment of the S/pol gene was cloned and 15 clones sequenced. BLASTn analysis showed all clones to contain an HBV-G specific insert (data not shown).
We report for the first time from the African continent clinical details of patients with HBV-G and the characterization of the genome. We identified 3 patients with HBV-G over a 9 year period from 2007-2015 in Cape Town, South Africa. All were MSM, aged between 30 and 50 years of age. Two patients were co-infected with HIV and one was also co-infected with HBV-A.
Liver biopsy in the co-infected patients showed moderate to severe fibrosis. Existing data, albeit conflicting, suggests that infection with HBV-G is associated with advanced fibrosis leading to a more rapid progression of liver disease, particularly in HIV co-infection [22,37]. A study by Calin et al. [38], comparing HIV patients co-infected with either HBV-G or HBV-non G demonstrated no such association and suggested the co-infection with other hepatitis viruses and lower CD4+ cell counts may play a more important role. Further, chimeric SCID mice expressing human hepatocytes show increased fibrosis when infected with HVB-G and HBV-A than in mono-infected mice [39]. In our report, the HIV coinfected patients had relatively low CD4+ cell counts of 319 and 347 cells/ mm3 , while one (P-2014) was also co-infected with HBV-A.
HBV-G is unable to express HBeAg due the presence of 2 stop codons at position 2 and 28 resulting in a truncated and therefore non-functional precore gene [10,13,40]. All 3 patients in this study were HBeAg-positive, indicating that a co-infecting HBV or a recombinant virus needs to be present to provide the precore protein, the precursor to HBeAg, in trans. HBV-A was found circulating with HBV-G in patient P-2014, but no evidence of another HBV genotype could be detected in patient M-2015 using the multiplex PCR method, which has a detection limit of 102 -103 for genotypes HBV-A, HBV-B, HBV-C, HBV-D and HBV-G and 104 and 105 for genotypes HBV-F and HBV-E, respectively [31]. Neither did cloning and sequencing of the pol/S gene of patient M-2015 show any other HBV genotype. Studies by Komatsu et al. [41] and Gong et al. [42] showed that sequencing of 16 or 20 clones resulted in the detection of 5 and 6% of a minor HBV mutant population, respectively. However, Araujo et al. [15] screened over 400 clones to detect 4 non HBV-G clones. Further, it is possible to calculate with 95% certainty the ability to detect a minority population using the formula 1-10^ [10log[0.05]/nclones] [13]. Thus sequencing of only 15 clones may have been inadequate to detect a minor non HBV-G population circulating below 10%. In this regard the use of next generation sequencing technologies which are capable of detecting low level viral populations circulating at >10% may have been a more suitable approach [43]. The INNO-LiPA HBV Genotyping line probe assay may also have detected a mixed infection [44]. As no recombination events were detected in the HBV-G of patient M-2015 by boot scanning of the complete genome, HBeAg could not have been synthesized by a recombinant virus. These results would suggest that, if present, the co-infecting virus is circulating at levels below the detection limits of the assays used. In the third patient, L-2007, there was no more sample available to determine if co-infection with another HBV genotype was present.
HBV-G mono-infections have been rarely described. A transient mono-infection was reported in a Dutch blood donor negative for HBeAg, HBeAb and HBsAg with a low viral load of 288 IU/ml [13]. A German apheresis donor with transient detection of HBsAg and negative serological markers for HBeAg and HBeAb, transmitted HBV-G to 2 platelet recipients [45]. A recent study by Cornelissen et al. [46] identified 5 possible cases of HBV-G mono-infection in MSM patients in Amsterdam. Although no other genotype could be amplified, based on the HBeAg status only 1 patient could be considered truly infected with only HBV-G. It is unlikely that our patients were mono-infected due to the presence of HBeAg.
At the molecular level the genomes contained 2 stop codons in the precore gene at positions 2 and 28 which prevents the synthesis of HBeAg [10,13,40]. All HBV-G isolates harbour the double A1762T transversion and G1764A transition mutations in the BCP which have been show in in vitro and in vivo studies to decrease the transcription of precore mRNA resulting in reduced expression and secretion of HBeAg [47]. In other genotypes, this double 1762T/1764A mutation usually develops at a late stage in HBV chronic infection [48]. Interestingly, patient P-2014 had the wild type 1764G at this position. Isolates with only 1762T or 1764A mutations are rarely reported and the appearance of 1764A is considered an intermediate in the development of the double 1762T/1764A mutation, as the 1764A mutation usually precedes the generation of the second 1762T mutation [47-49]. These 2 mutations also cause non synonymous changes in the overlapping X gene resulting in amino acid K130M and V131I substitutions which may influence this multifunctional regulatory protein [48]. Insertion of 36nt near the translation initiation codon of the core gene, which is unique to all HBV-G isolates, adds 12 amino acids (DRTTLPYGLFGL) immediately after the initiating methionine. This insert also affects the secondary structure of the encapsidation signal (Ɛ) which is required to direct the pre genomic RNA into core particles for replication [34]. The first 3 nucleotides (TAG) slightly alter the base pairing of the lower stem structure of the Ɛ signal [11]. There are conflicting reports as to the effect of this 36nt insert on viral replication, core protein expression and virion secretion. Li et al. [34] showed in vitro that HBV-G alone was not only replication competent, but also more efficient in core protein expression and secretion of viral particles with a more mature genome. Although more recent studies by Cotelesage et al. [50], Gutelius et al. [11] and Sakamoto et al. [51] have confirmed the overproduction of core protein, but these studies demonstrated that viral replication was impaired and the insert caused structural changes to the larger core protein hindering envelopment and viral secretion. A possible reason why only HBV-G has this insert is that it sustains core production required for particle assembly in response to low replicative capacity resulting from low pg RNA levels [11].
Gene expression can be regulated by DNA methylation which is associated with CpG dinucleotides grouped in clusters called CpG islands [52]. When these islands are located in promoter regions hypermethylation can occur leading to gene silencing at the transcription level [52]. HBV has 4 promotors, cp, sp1, sp2 and xp, as well as enhancer I and II which have stimulatory effects on these promotors [48]. Depending on the HBV genotype, the genome has 2-3 conventional islands, CpG I, II and III, and a further 3 novel CpG islands have been found in some HBV-B,HBV-C, HBV-D, HBV-F and HBV-H isolates [53,54]. HBV-G has only CpG islands II and III, with a gap or “split” found in CpG II in 92% of the 40 full HBV-G genomes analyzed in this study, confirming the data of Zhong et al. [54]. This “split” phenotype was also observed in genotypes A, D, E, and F at rates of 2%, 30%, 29% and 50%, respectively [54]. How this “split” occurs and the biological and clinical significance is not clear. CpG island II is located in an important regulatory region which plays a role in RNA transcription, protein production and viral replication [48]. The CpG island III encompasses the P gene start site and overlapping 3’end of the core gene. The distribution and number of the CpG islands in the different HBV genotypes may influence methylation-mediated gene regulation and thereby affect clinical outcomes.
Phylogenetic analysis of the complete HBV-G genomes of 2 patients, P-2014 and M-2015, found that they clustered together and were phylogenetically very similar to all HBV-G references sequences in the GenBank database. The genetic homology of the these 2 South African isolates with 4 unique nucleotide changes in common would suggest a possible shared source, unlike L-2007 which is phylogenetically more distant, forming a distinct branch of the HBV-G tree. The phylogenetic tree and sequence similarity could not provide information as to whether HBV-G was acquired locally or not.
Lindh [27] suggested that HBV-G may be of African origin as it is most closely related to HBV-E, which is considered an African virus being prevalent in West and Central Africa and only found in other parts of the world in HBV carriers of African origin [7]. These 2 genotypes are the least divergent from each other at 11% and share an almost identical 30 nt segment in the pre S region and a 3 nt deletion at the amino end of this protein [27,36] . Therefore HBV-G may have acquired the 30 nt fragment through a recombination event with HBV-E [27]. Further, both genotypes have a very low degree of genetic diversity and each forms a single monophyletic group with no sub genotypes, indicating a recent evolutionary history in humans. Lindh [27] has proposed that these common molecular features may indicate a geographical origin in Africa. To date no HBV-G or HBV-G/E recombinants have been found in patients of African ancestry and the 3 patients described in this study, were not of black African ethnicity
Download Provisional PDF Here
Aritcle Type: Research Article
Citation:Smuts H, Sonderup M, Gogela N, Spearman CW (2017) Hepatitis B Virus Genotype G: First Report of Complete Genomic Analysis from the African Continent. J Emerg Dis Virol 3(2): doi http://dx.doi.org/10.16966/2473-1846.130
Copyright: © 2017 Smuts H, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access