Table 1: Demographic and clinical characteristics of primary Sjögren syndrome patients and healthy individuals

Silvia Reina1,2 Cecilia Pisoni3 Alicia Eimon3 Carolina Carrizo3 Sabrina Ganzinelli1,2 Roberto Arana3 Enri Borda1,2*
1 Pharmacology Unit, School of Dentistry, University of Buenos Aires, Buenos Aires, Argentina*Corresponding author: Enri Borda, M.D., Ph.D., Pharmacology Unit, School of Dentistry, University of Buenos Aires,Marcelo T de Alvear 2142 – 4 “B”, 1122 AAH. Ciudad Autónoma de Buenos Aires,Argentina, Tel: +54-11-4964-1276; E-mail: enri@farmaco.odon.uba.ar
Aims
This paper investigates the action of M3 muscarinic acetylcholine receptor antibody present in serum from patients with Sjögren syndrome (SS).
Methods
Enzyme-linked immunoabsorbent assay (ELISA) was performed in the presence or absence of different enzymatic and specific receptors’ antagonist drugs. The levels and the generation of PGE2 , 6-keto-PGF1α and cyclic AMP (cAMP) in rat submandibular gland acini’s preparations and in serum from pSS patients were measured in the presence of pSS IgG anti M3 peptide. COX-2 mRNA gene’s expression at Real Time PCR was done in acini’s preparations from rat submandibular gland in the presence of the autoantibodies alone or once previously incubated with different inhibitors.
Results
In this study, we show that the activation of M3 mAChR of rat submandibular gland acini’s preparation triggers an increment both in the production of COX-2 mRNA gene’s expression and in the production of PGE2 and 6-keto-PGF1α. These phenomena are accompanied by an increment in the production of cAMP in the acini’s preparation and do not affect COX-1 mRNA’s levels. Both prostanoids are augmented in the sera of pSS patients as compared with healthy individuals.
Conclusions
The present study suggests a complex interplay between different factors involved in adaptativa autoimmunity in pSS patients at the level of exocrine glands. The presence of pSS IgG anti M3 peptide, the enhancement of COX-2 mRNA gene’s expression and the increment in the generation of PGE2 and 6-keto-PGF1α abolished by M3 specific cholinergic antagonist, could provide evidence of a link between autoimmunity and the submandibular gland parasympathetic system in the course of Sjögren’s syndrome. This evidence is further supported by an increment in the production of AMP cyclic nucleotide (cAMP), and the subsequent induction of desensitization, internalization and/or intracellular degradation of the glandular M3 mAChR displayed by the cholinergic autoantibody. All of these statements cited above, are responsible for xerostomy, xerophthalmia and other parasympathetic symptoms observed in SS patients.
Autoantibodies; Sjögren’ syndrome; PGE2 ; PGI2; cAMP; COX-2 mRNA
Primary Sjögren’s syndrome (pSS) is a common chronic autoimmune disease characterized by the inflammation of exocrine glands and the clinical signs of xerostomia and keratoconjuntivitis sicca. A combination of environmental, genetic and possibly hormonal factors leads to the dysregulation of the glandular epithelium, mononuclear cell infiltration and abnormal lymphocyte activation and proliferation [1,2]. Alter humoral autoimmune responses, B cell hyperactivity and autoantibodies production are the hallmarks of pSS [3-5].
We have reported the presence of autoantibodies against rat salivary and lacrimal glands M3 muscarinic acetylcholine receptors (mAChR M3 ), which trigger parasympathetic-receptor-mediated biological effects [6-9]. We also demonstrated that they are able to recognize a synthetic peptide corresponding in amino acid sequence to the second extracellular loop of the human M3 mAChR. An isolated fraction of pSS IgG rich in anti M3 peptide antibodies could reproduce the effects of the corresponding whole immunoglobulin.
These facts suggest a prominent role of the anti M3 peptide antibody in the mAChR-mediated effects of total pSS IgG.
Other known facts are that the synthetic M3 peptide involved selectively suppresses the biological effects of SS anti M3 peptide autoantibody and the corresponding total IgG. The neurotransmitter mAChR autoantibodies coexist with the presence of other important autoantibodies to Ro/SS-A (Ro52, Ro60) or La/SS-B [10-12]. These autoantibodies are non-organ specific. Their role in the pathogenesis of SS has not been understood yet.
This supports the view that the second extracellular loop is not only the main immunogenic region of the receptor but can be considered essential for the biological action of these autoantibodies.
In this context, it is important to notice that antinuclear antibodies (ANA) are frequently found together with the rheumatoid factor (RF) and anti centromere antibody (ACA) in patients with pSS in the early stages of the disease and at a younger age. These autoantibodies are not specific to SS, but show a local response to autoantigens derived from salivary glands aggressors, which produce these autoantibodies at the level of the local system [13]. Prostaglandins (PGs) are implicated in normal cellular processes as well as in pathophysiological conditions such as inflammation [14,15]. PGE2 and PGI2 are synthesised by cyclooxygenase (COX) and prostaglandin and prostacyclin E and I synthases in vivo. The two enzymes catalyse the reaction of transformation of arachidonic acid (AA) through PGH2 into PGE2 and PGI2. The two isoforms of COX (COX- 1 and COX-2) and PGE2/ PGI2 have been identified: In general, COX- 1 is constitutively expressed in almost all tissues and have haemostatic effects, whereas inflammatory processes lead to an increased expression of COX-2 [16] in response to inflammation; the increased expression of COX-2 provokes, in turn, an increment in the generation of PGE2 and PGI2; PGE2 has been shown to be part of the signalling events involved in M3 mAChR activation [17,18].
Accordingly, the aim of our work is to investigate if the inflammatory process provoked by the M3 mAChR IgG of sera of pSS patients in rat submandibular gland and the following increased expression of mRNA COX-2 and production of PGE2 and PGI2 are associated with the presence of M3 mAChR IgG.
The study protocol complied with the tenets of the Declaration of Helsinki and accomplished with the rules established by the Ethics Committee of the University of Buenos Aires (Buenos Aires, Argentina). All subjects provided written informed consent.
A 25-mer peptide (K-R-T-V-P-D-N-Q-C-F-I-Q-F-L-S-N-P-A-V-TF-G-T-A-I) corresponding to the sequence of the second extracellular loop of the human M3 mAChR was synthesized by Peptide Genetic Research Company (Livermore, CA, USA) as previously described [6]. DuP697 5 × 10-6 M (5-bromo-2-(4-fluorophenyl)-3-(4-(methylsulfonyl) phenyl-thiophene); RO3144794 5 × 10-8 M (R-3-(4-fluoro-phenyl)-2-[5- (4-fluoro-phenyl)-benzofuran-2-ylmethoxycarbonylamino]-propionic acid); PF-04418948 2 × 10-9 M (1-(4-fluorobenzoyl)-3-([6-methoxy-2- naphthyl) oxy] [methyl] azetidine-3-carboxylic acid) were from Tocris Cookson (Ellisville, MO, USA). Stock solutions were freshly prepared in the appropriate buffers and the drugs were diluted in water to achieve the final concentrations stated in the text.
Male Wistar rats weighing 250-300 g from the Pharmacologic Bioterium School of Dentistry, University of Buenos Aires) were used throughout. The animals housed in standard environmental conditions were fed with a commercial pellet diet and water ad libitum. For surgical removal of submandibular glands, the animals were killed with an overdose of an i.p. ketamine/xylazine mixture (100 and 16 mg/kg respectively). The experimental protocol followed the Guide to The Care and Use of Experimental Animals (DHEW Publication, NIH 80-23).
Submandibular gland acini’s were prepared from adult female Wistarstrain rats. Animals were used according to “The Guide to the Care and Use of Experimental Animals” (DHEW Publication, NIH 80-23). Glands were dissected away from fat, connective tissue and lymph nodes and immersed in a tissue chamber containing Krebs-Ringer-bicarbonate (KRB) solution gassed with 5% CO2 in oxygen and maintained at pH 7.4 and 37°C. All subsequent steps were performed at 4°C. Submandibular glands were minced and incubated in KRB supplemented with 10 mM HEPES and 5.5 mM glucose (KRB-HEPES) and 0.5% bovine serum albumin (BSA), pH 7.4, containing collagenase (150 U/ml). Gland lobules were subjected to gentle pipetting. The preparation was then filtered through nylon mesh (150 µm pore size) and the acini’s were pellets with a 2 min centrifugation at 50 g. The pellet was then washed twice by centrifugation (50 g for 2 min) through a 4% BSA solution made with KRB-HEPES buffer. The dispersed acini’s+ were allowed to recover for 30 min in 5 ml fresh KRBHEPES buffer containing 0.5% BSA [19].
The subjects of this study were 28 pSS anti-Ro/SSA positive patients and 25 healthy volunteers, all female, aged 39-54 years. They were selected from the metropolitan area of Buenos Aires (Table 1). The diagnosis of pSS fulfilled the criteria described by Vitali et al. [20] and was given by means of a positive biopsy with a score focus of 3.8 ± 0.07.
The serum IgG fraction from patients with pSS and from normal individuals (control) was isolated using protein G affinity chromatography as described elsewhere [21]. Briefly, sera were loaded onto the protein G affinity column (Sigma-Aldrich, St Louis, MO, USA) equilibrated with 1 M Tris-HCl (pH 8.0) and the columns were washed with 10 volumes of the same buffer. The IgG fraction was eluted with 100 mM glycine-HCl, pH 3.0, and immediately neutralized. The concentration and purification of IgG were determined using a radial immunodiffusion assay
Anti-Ro-SSA procedure: Saline-soluble extractable nuclear antigens (ENA) were obtained from human spleen in phosphate buffered saline (PBS) for anti-Ro. Patient sera were tested undiluted and diffusion was carried out at room temperature in a humidified chamber for 48 hours. Precipitin lines were identified by comparison with reference sera. ELISAs for total anti-Ro (60 kD and 52 kD Ro-proteins) was performed with a commercial Kit based on purified antigens (Orgentec Diagnostika, Mainz, Germany) and the assays were carried out according to the manufacturer’s protocols on an automated ELISA instrument (Radim, Pomezia RM, Italy). Values greater than 25 UI/ml was considered positive (Table1).
Anti-M3 peptide IgG procedure: The IgG fraction from 28 patients with pSS and 25 healthy subjects was independently subjected to affinity chromatography on the synthesized peptide covalently linked to AffiGel 15 gel (Bio-Rad, Richmond, CA, USA) as described by Reina et al. [8]. Briefly, the IgG fraction was loaded onto the affinity column equilibrated with PBS. The non-peptide fraction was first eluted with the same buffer. Specific anti-peptide antibodies were then eluted with 3 M KSCN and 1 M NaCl, followed by immediate extensive dialysis against PBS. The IgG concentration of non-anti-peptide antibodies and specific antimuscarinic receptor peptide antibodies was determined by a radial immunodiffusion assay. Their immunological reactivity against muscarinic receptor peptides was evaluated by using ELISA. The concentration of the affinity-purified anti-M3 peptide IgG (1×10-8 M) increased optical density (mean OD ± SEM, 2.4 ± 0.2). The non-anti-M3 peptide IgG fraction eluted from the column showed OD values (0.27 ± 0.06) similar to those of normal IgG from healthy individuals taken as control (0.26 ± 0.05). The normal IgG fraction purified by affinity column chromatography gave a negative result (0.30 ± 0.03) (Table1). ELISA was performed as described previously [22].
Table 1: Demographic and clinical characteristics of primary Sjögren syndrome patients and healthy individuals
Total RNA was extracted from rat submandibular gland acini’s using Trizol reagent (Invitrogen Life Technologies, CA, USA) according to the manufacturer´s protocols as previously reported [23]. One μg of total RNA was treated with DNAase Kit FERMENTAS, St. Leon-Rot, Germany) and subsequently reverse-transcribed into cDNA using Iscrip cDNA Synthesis Kit (Bio-Rad Laboratories, CA, USA) according to the manufacturer`s protocols. One hundred nanogram of cDNA was used for the reaction mixture. The amplification of COX-2 and glyceraldehydes 3-phosphate dehydrogenase (GAPDH) was performed using iTaqTM Fast SYBR® Green Supermix with ROX (Bio-Rad). The couples of primers that resulted from using the 7900HT Fast Real-Time PCR system (Applied Biosystems, CA, USA) were as follows: COX-2-: 5′GCTCAGCCATACAGCAAATCC; rev: 5′CCAAAATTAAC; GAPDH: 5′TCACCAGGGCTGCTTTTAAC; rev: 5′GACAAGCTTCCCGTTCTCAG. COX-2 mRNA expression were normalized with GAPDH levels. Gene expression assays were performed by relative quantification with comparative cycle threshold using ABI Prism SDS 2.-1 software (Applied Biosystems) and the results were expressed as per cent of changes.
Tissue (submandibular gland acini’s) PGE2 and 6-keto-PGF1α were measured by ELISA, which was carried out according to the manufacturer’s protocols (Biotrack Enzyme Immune Assay System, Amersham Bioscience, Piscataway, NJ, USA). The OD cut-off values for pSS serum PGE2 were 42.6 ng/L and 6-keto-PGF1α 64.2 ng/L; the OD cut-off values for gland acini’s PGE2 were 0.5 ng/ml and 6-keto-PGF1α 0.62 ng/ml (basal control). All gland acini’s samples were frozen promptly after collection and kept at -80°C until used for PGE2 and 6-keto-PGF1α determination. The result in serum is expressed as ng/L and in submandibular gland acini’s in ng/ml.
To determine cyclic AMP (cAMP) levels tissues (submandibular gland acini’s) were incubated in 1 ml of Krebs Ringer Bicarbonate (KRB) for 30 min. After incubation, submandibular gland acini’s were homogenized in 2 ml of absolute ethanol and centrifuged at 6000 g for 15 min at 4°C. Pellets were then re-homogenized in ethanol–water (2:1) and supernatants were collected and evaporated to dryness. The residue was dissolved in 400 μl of 50 μM sodium acetate buffer, pH 6.2 and aliquots of 100 μl were taken for nucleotide determination using a radio immunoassay procedure with a 3H-cAMP-RIA kit from Dupont New England Nuclear (Boston, MA, USA).
The Student’s “t” test for unpaired values was used to determine the level of significance. In case multiple comparisons were necessary after analysis of variance the Student-Newman-Keuls test was applied. Differences between means were considered significant if P<0.05.
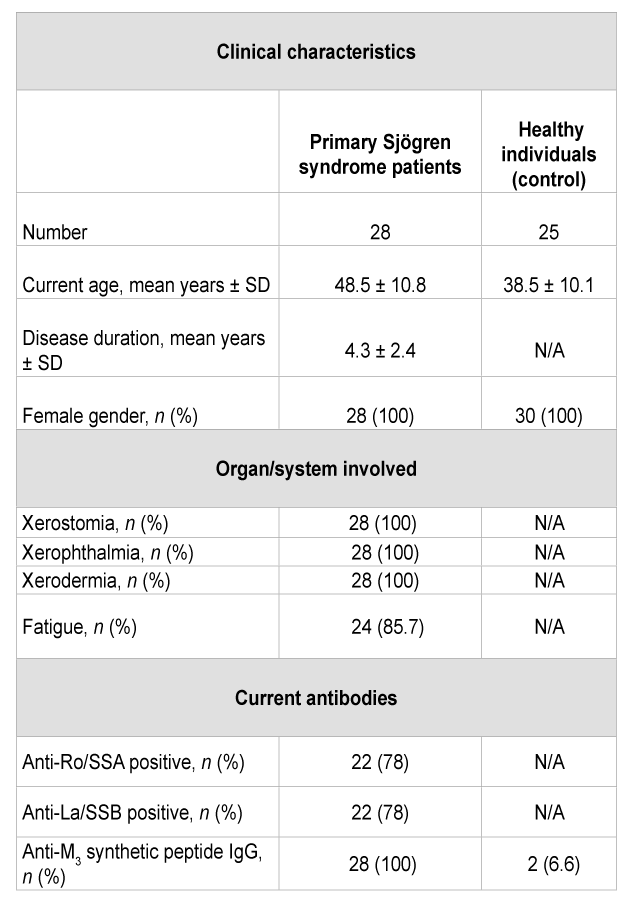
To determine the effect of pSS IgG anti M3 peptide on gland acini’s we analyzed the time-course of COX-2 mRNA expression by Real Time-PCR (Figures 1A and B). At the sixth hour presence of pSS IgG anti M3 (1 × 10-8 M), COX-2-mRNA was significantly (P<0.001) increased versus normal IgG anti M3 , which is ineffective in this studied system (control). When the autoantibody was incubated in the presence of DuP697 (1 × 10-6 M) [a specific COX-2 antagonist] under the same experimental conditions, it abrogated the increment of COX-mRNA’s expression in a significant manner (P<0.01). However, when the experimental protocol was assessed in the presence of M3 mAChR specific antagonist J104127 (1 × 10-8 M) and/or synthetic M3 peptide (5 × 10-6 M) the change in COX-2 mRNA expression by the pSS IgG anti M3 did not reach statistical significance (Figure 1C). COX-1 mRNA expression did not change significantly in our preparation system.
As shown in Figure 2, the dose-response concentration curve of PGE2 (A) and 6-keto-PGF1α (C) in the presence of pSS IgG anti M3 on submandibular gland acini’s increased the production of both prostanoid, reaching the maximal action when the antibody is at a concentration of 1 × 10-8 M. The increment in the generation of both prostanoids is abrogated, reaching values similar to basal ones, when the tissue preparations are incubated with prostanoid antagonists PF-04418948 2 × 10-9 M (Figure 2A) and RO3244794 5 × 10-8 M (Figure 2C) for PGE2 and 6-keto-PGF1α respectively, and in the presence of synthetic M3 peptide 5 × 10-6 M (Figures 2A and 2C). Normal (n) IgG anti M3 peptide was ineffective in our study system (Figure 2 histogram B and D).
Synthetic M3 peptide 5 × 10-6 M, PGE2 5 × 10-6 M and 6-keto-PGF1α 5 × 10-6 M were accompanied by modification of cAMP production associated positively with the increased amount of both prostanoids (Figure 3 and Table 2).
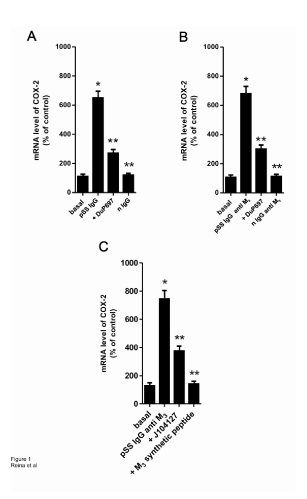
Figure 1: Bar graph depicts the mRNA expression of cyclooxygenase-2 (COX- 2) in rat submandibular gland acini’s. The acini’s from eight rats were treated during 3 hours with or without (basal) pSSIgG (1x10-7 M) or IgG anti M3 peptide (1x10-8 M) or n IgG anti M3 peptide from healthy individuals (normal: n IgG) and primary Sjögren`s syndrome patients (pSS patients) respectively. In inhibitory experiments, COX-2 inhibitor (DuP697 5x10-8 M), M3 specific mAChR antagonist (J10427 1x10-9 M) and M3 synthetic peptide (1x10-5 M) were added to acini’s 30 minutes before pSS IgG or pSS IgG anti M3 treatment. The COX-2 mRNA was extracted using quantitative real-time polymerase chain reaction and the levels of COX-2 were measured. Values are mean ± SEM expressed as per cent of change of 5 experiments in each group performed by duplicate. *P<0.0001 versus basal (acini’s alone); ** P<0.001 versus pSS IgG or pSS IgG anti M3 .
In Figure 3A it can be seen that the production of cAMP reaches maximal increment at 1 × 10-8 M pSS IgG anti M3 peptide. This stimulatory effect was almost abolished by pre-treating submandibular gland acini’s with J104127 1 × 10-8 M and/or synthetic M3 peptide 5 × 10-6 M. The normal IgG anti M3 peptide gave negative results (Figure 3A).
To ascertain if this cAMP increment is responsible for the augmented generation of PGE2 and 6-keto-PGF1α in our preparation, we study the action of both prostanoids on the production of this nucleotide. Figure 3B and Figure 3C shows that both prostaglandins are able to increase cAMP production, whereas the selective prostanoids antagonists (PF-04418948 for PGE2 and RO3244794 for 6-keto-PGF1α) blunt the stimulatory action provoked by the prostaglandins.
The distribution of PGE2 and 6-keto-PGF1α was studied in serum of pSS patients and in healthy individuals. The scatter grams of Figure 4 show both 6-keto-PGF1α’s (A) and PGE2 ’s (B) concentration in serum from pSS patients and healthy individuals (control). The concentration of 6-keto PGF1α and PGE2 in serum of pSS patients was two standard deviations higher than that in normal individuals (P<0.001).
In this study, we show that the activation of M3 mAChR of rat submandibular gland acini’s preparation triggers an increment in the production of COX-2 mRNA gene’s expression together with an increment in the production of PGE2 and 6-keto-PGF1α. These phenomena are accompanied by an increment in the production of cAMP in acini’s preparation without affecting COX-1 mRNA levels.
Positive regulation by cholinergic agonists has been described in different tissues [24-27] including rat submandibular gland [28]. We observed that such stimulation was due to COX-2’s activation through M3 mAChR glandular acini’s activation: this increment was prevented by the specific blockade of this enzyme [29], by the cholinoceptor antagonist agent [30] and by M3 synthetic peptide [16]. However, the rat submandibular gland constitutively expressed COX-1 [31]. In most cells, COX-1 mediates physiological responses such as the modulation of the autonomic nervous system, whereas, COX-2 mainly plays a role in inflammation, infection and cellular proliferation [22]. In our case, the autoantibody from sera of pSS patients was able to stimulate the COX-2 mRNA’s expression. This was the case due to the fact that when this pSS IgG anti M3 peptide binds to, and activates, the M3 mAChR receptor present in the membrane of gland acinar cells; it provokes an inflammatory process, in which COX- 2 is expressed [23]. COX-1’s expression did not change significantly in the rat submandibular gland acini’s preparations in the presence of the cholinergic autoantibody.
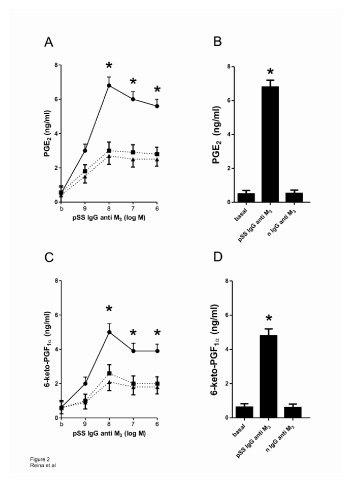
Figure 2: A: Concentration-response curve of pSS IgG anti M3 peptide alone (●) or in the presence of PF-04418948 2x10-9 M (■) and J104127 1x10-8 M (▲) on the generation of PGE2 ; B: Histogram shows in comparative form in acini’s preparations alone, the action of the maximal concentration (1x10-8 M) of pSS IgG anti M3 peptide in comparison to normal (n) IgG anti M3 peptide on the generation of PGE2 ; C: Concentration-response curve of pSS IgG anti M3 peptide alone (●) or in the presence of RO3244794 5x10-8 M (■) and J104127 1x10-8 M (▲) on the generation of 6-ketoPGF1α; D: Histogram shows in comparative form in acini’s preparations alone, the action of the maximal concentration (1x10-8 M) of pSS IgG anti M3 peptide in ○Values are mean ± SEM of 5 experiments in each group performed by duplicate; *P<0.001 versus values in dashed line; **P<0.0001 versus basal (b) and nIgG anti M3 values.
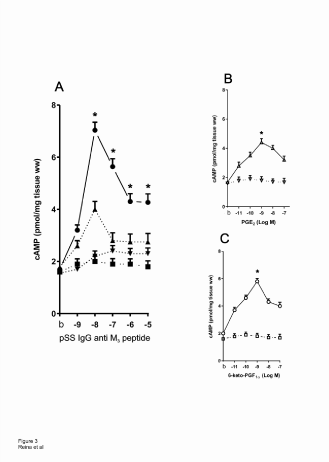
Figure 3: A: Concentration-response curve of pSS IgG anti M3 peptide alone (●) and in the presence of J104127 1x10-8 M (▲) and M3 synthetic peptide 1x10-5 M (▼) on submandibular gland acini’s preparation cAMP production. B and C: As control, concentration-response curve of PGE2 alone (∆) and PF-04418948 2x10-9 M plus PGE2 (∆) and 6-ketoPGF1αalone (○) and RO3244794 5x10-8 M plus 6-keto-PGF1α (□) on submandibular gland acini’s preparation cAMP production. Different points represent the media ± SEM of 4 experiments performed by duplicate in each concentration. *P<0.0001 versus different inhibitors (dotted/dashed lines in A, B and C).
On the other hand, pSS IgG anti M3 peptide has a capacity to stimulate the production of PGE2 and 6-keto-PGF1α in concentration-response curve fashion in gland acini’s preparations. However, the stimulatory action of these autoantibodies is inhibited by M3 mAChR’s specific antagonist [30], by PGE2 ’s specific antagonist [31], 6-keto-PGF1α‘s antagonist [32] and M3 synthetic peptide [16]. This shows that the action of these autoantibodies on acini’s glandular preparations act as an inducer of the augmented generation of both prostanoids. The prostanoids, in turn, are able to maintain chronic inflammatory processes at the level of submandibular gland in the chronic course of pSS.
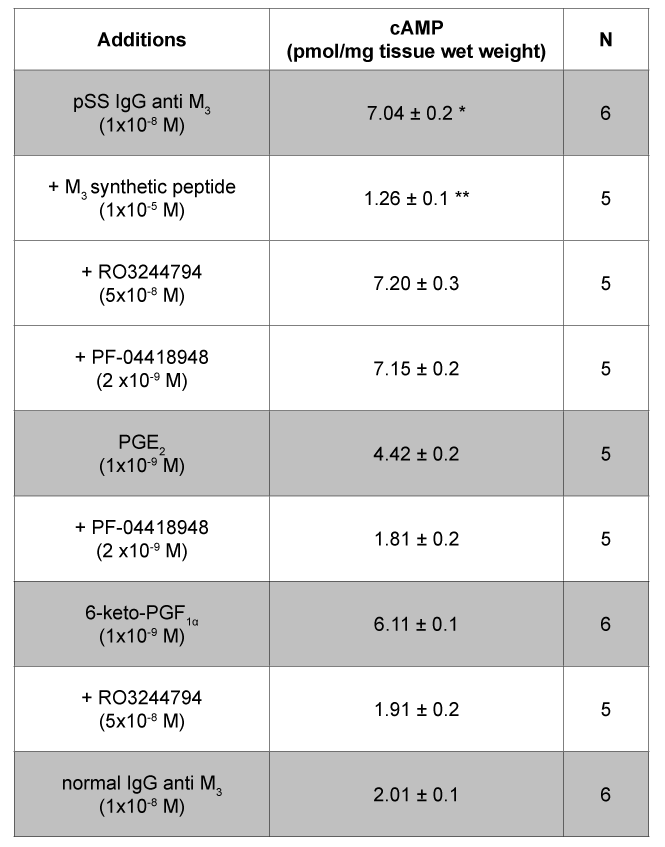
Table 2: Tissue production of cAMP in rat submandibular gland acini in the presence of pSS IgG anti M3 , PGE2 and 6-keto-PGF1α Values are mean ± SEM of n experiments performed by duplicate. * P<0.0001 versus normal IgG anti M3 ; ** P<0.0001 versus pSS IgG anti M3 , PGE2 and 6-keto-PGF1α. PF-04418948 (PGE2 antagonist) and RO3244794 (PGI2 antagonist) did not have any effect per se in the system.
It has long been known that prostanoids can be generated by most cells in response to mechanical, thermal or chemical injuries, and/or inflammatory process [33]. Accordingly, the proinflammatory effects of PGE2 and PGI2 have been studied widely in numerous models of inflammation [34]; PGE2 and PGI2’s action, are also seen as being able to contribute to the vasodilatory blood flow in all tissues acting as proinflammatory cytokine [35]. Both prostanoids can be considered to play a crucial role in the process of inflammation (occurring at the level of the exocrine glands, i.e., eye and salivary gland), which is itself responsible for the xerophthalmia and xerostomia present in pSS provoked by the cholinergic autoantibody
In all these pharmacological approaches the increment of COX-2 / PGE2 / PGI2 in the submandibular gland acini’s preparations through the action of pSS IgG anti M3 peptide is shown to be the result of the ability of this cholinergic autoantibody present in the sera of pSS patients to mobilize arachidonic acid from phospholipids. The presence of this autoantibody is shown to lead to the subsequent activation and increase of the COX-2 mRNA’s expression accompanied by a large amount of PGE2 / PGI2. Both prostanoid are thus shown to be responsible of the installation of the neuroinflammatory pathophysiological processes at the level of glandular salivary gland.
The increment in PGE2 and 6-keto-PGF1α is mediated by cAMP’s increased production, whereas the increased production of this nucleotide is inhibited by the prostanoids’ specific antagonists. In acini’s preparations pSS IgG anti M3 peptide is able per se to enhance cAMP’s production and this nucleotide is blunted by M3 cholinergic antagonist and M3 synthetic peptide. IgG anti M3 of normal individuals (control) is without effect in our system. When pSS IgG anti M3 peptide from sera of pSS patients bind to, and activate, rat submandibular gland acini’s M3 mAChR, the autoantibody leads to the activation of adenylate cyclase. What follows is an increased production of cAMP together with the increment of the inflammatory mechanism of the gland. This can be explained by the fact that the nucleotide ultimately makes the same contribution to the modulation of the gland apoptotic pathways as observed in SS [36].
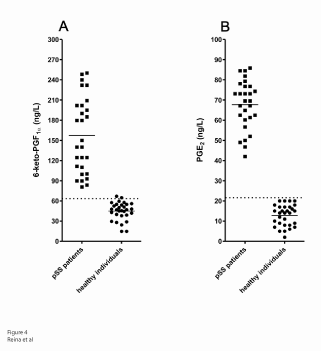
Figure 4: Detection of serum levels of 6-keto-PGF1α and PGE2 . Scatter gram showing the immune reactivity of serum levels of 6-keto-PGF1α (A) and PGE2 (B). Individual values for each serum from 28 pSS patients and 30 healthy individuals (normal). Dotted/dashed line, cut-off values 62.5 g/L and 25.2 g/L (mean values ± 3 SD for healthy individuals) for 6-ketoPGF1α and PGE2 respectively. Solid lines, median values expressed as ng/L: 157.30 ± 10.61 (pSS serum) and 44.89 ± 2.41 (normal serum) for 6-keto-PGF1α, P<0.0001; and 67.69 ± 2.98 (pSS serum) and 12.81 ± 0.95 (normal serum) for PGE2 , P<0.0001.
The present study suggests a complex interplay between different factors involved in adaptativa autoimmunity in pSS patients at the level of exocrine glands.
The presence of pSS IgG anti M3 peptide, the enhancement of COX- 2 mRNA gene’s expression and the increment in the generation of PGE2 and 6-keto-PGF1α - abolished by M3 specific cholinergic antagonist - could provide a link between autoimmunity and the submandibular gland parasympathetic system in the course of Sjögren syndrome. This view is further supported by the increment in the production of AMP cyclic nucleotide (cAMP). Another important fact in this concern is that the cholinergic agonist activity displayed by the cholinergic autoantibody could subsequently induce desensitization, internalization and/or intracellular degradation of the glandular M3 mAChR, resulting in xerostomy, xerophthalmia and other parasympathetic symptoms observed in SS patients.
This work was supported by grants from the Buenos Aires University No. 200200130100325BA.
The authors declare no conflict of interest.
Download Provisional PDF Here
Article Type: Research Article
Citation: Reina S, Pisoni C, Eimon A, Carrizo C, Ganzinelli S, et al. (2015) Changes in Cyclooxygenase- 2’s Expression, and PGE2’s and 6-keto-PGF1α’s Levels in the Presence of the Muscarinic Acethylcholine Receptor Antibody in Primary Sjögren Syndrome. Int J Dent Oral Health 1 (3): doi http://dx.doi.org/10.16966/2378-7090.115
Copyright: © 2015 Reina S et al. This is an openaccess article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Publication history:
All Sci Forschen Journals are Open Access