Introduction
Ankylosing spondylitis is a chronic systemic inflammatory disorder
that primarily involves the sacroiliac joints and axial skeleton. It is part
of the group of spondyloarthropathies (SpA), which is one of the most
frequently occurring groups of inflammatory rheumatic disorders
[1]. Patients are typically between ages 20 and 40 [2] and the most
common symptoms are fatigue, inflammatory back pain, and peripheral
enthesitis and arthritis; extra-articular manifestations, such as uveitis and
diseases involving the pulmonary, cardiovascular, renal, neurological, or
gastrointestinal systems, may also be present [3-5].
Although the etiology of AS is not completely understood, it is clear
that both environmental and genetic factors contribute to the disease. As
evidence of the importance of genetics, the class I major histocompatibility
complex molecules (MHC-I) appear to play the most significant role
in AS susceptibility [6]. The strongest association is with HLA-B27;
approximately 90% of AS patients are HLA-B27 positive [7]. In fact,
HLA-B27 imparts the largest relative risk of developing an autoimmune
disease of any MHC-linked autoimmune disease. HLA-B27 was the first
predisposing allele found in all forms of SpA more than 40 years ago [8].
A significant association was also found in a very small cohort, between
AS and B*14, where 62.5% of the AS patients were HLA-B14+, but only 2%
of the healthy donors carried this allotype [9]. Apart from HLA-B27 and
HLA-B14, -B60 (a split antigen of -B40) and -B38 and -B39 (split antigens
of -B16) have also been linked to AS [10].
The fact that only 1-2% of the HLA-B27 positive population develops
AS [1,11], suggests that other factors also contribute to the pathogenesis
of AS. Erap1, a gene involved in MHC-I antigen presentation – the process
in which peptides are presented to CD8+ T cells – has been implicated,
which suggests that the MHC-I pathway is highly important in AS
pathogenesis. This makes it likely that other genes involved in the MHC-I
pathway could be important in the susceptibility of the disease, as well.
Also, genome-wide association studies have implicated over 30 genes in
susceptibility to AS [12]. Non-MHC-I genetic factors, such as IL-1A, IL-
23R, also contribute to AS susceptibility [13] but, in this review, I will
mainly focus on how MHC-I presentation contributes to the pathogenesis
of AS. Also, studies of identical twin-pairs revealed a high concordance
rate among siblings, but it is not 100% [14]. Therefore, environmental or
epigenetic factors must contribute. Among these, the microbiota has been
also recently involved as another important factor in the susceptibility to
the AS.
This review summarizes the extensive literature on how the MHC-I
molecule, antigenic peptides, other antigen presentation machinery,
and several different pathogens can all influence the development of AS,
together with other factors not involved in antigen presentation. This
review also evaluates the different hypotheses (Figure 1), which have
been proposed as an attempt to explain the mechanisms underlying the
association between HLA-B27 and AS pathogenesis. Individually, no
single molecule, factor or pathogen can take the credit for causing AS – it
is clearly a multi-factorial disease.
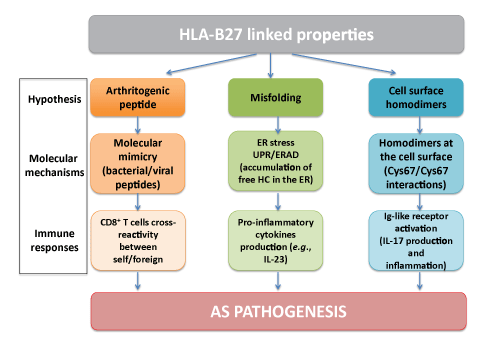
Figure 1: Flowchart showing the different hypotheses explaining the association with AS: HLA-B27 is a peculiar molecule with intrinsic properties
that have led to different hypotheses trying to explain the association of this MHC-I molecule with AS: As peptide presenting molecule, HLA-B27 has
the potential to present arthritogenic peptides. HLA-B27 tends to misfold in the ER and free HC tends to accumulate, leading to ER stress and the
UPR/ERAD responses and production of pro-inflammatory cytokines. Also, HLA-B27 can form cell surface homodimers, which can be recognized by
Ig-like receptors, followed by the stimulation of IL-17 production, leading to inflammation and, therefore pathogenesis. A detailed explanation of these
hypotheses is given in the first sections of this review.
HLA-B27
HLA-B27: The good, the bad and the ugly
In 1973, the discovery of a correlation between HLA-B27 and AS was
the first time an inflammatory disease was shown to be associated to a HLA
haplotype [7]. Since then, a number of other HLA-B27 syndromes have
been discovered, including acute anterior uveitis, reactive arthritis (ReA),
inflammatory bowel disease, and psoriatic arthritis. Yet, inheritance of
HLA-B27 is not exclusively bad. In fact, several studies have shown that
HLA-B27 has a protective role in human immunodeficiency virus
(HIV) and hepatitis C virus (HCV) infections, where possession of
HLA-B27 associated strongly with the absence of long term progression
in HIV infection and spontaneous clearance of HCV [15,16]. HLAB27-mediated
protection is probably a combination of multiple viral
and immunologic mechanisms, some of which may also be involved in
AS pathogenesis.
HLA-B27 Subtypes: Differential peptide specificity and
association to AS
HLA-B27 is an MHC-I molecule whose principal function is to present
peptides, mostly from endogenous proteins, at the cell surface for CD8+
T cells. To date, close to 150 subtypes of HLA-B27 have been identified
(http://www.ebi.ac.uk/ipd/imgt/hla/allele.html). The subtypes differ
in their peptide-binding specificity, which is defined by the peptidebinding
groove of this MHC-I molecule. The peptide-binding groove is
arranged by the α1α2 and domains of MHC -I: it is high polymorphic
[17] and consists of six side pockets (A-F) [18,19] that accommodate the
bound peptide’s side-chains [17]. Of particular importance are pocket B,
which accommodates the side chain of residue 2 (P2), pocket F, which
accommodates the C-terminal residue (usually P9) and pockets D and E
which bind the side chains of residues 3 and 7 of the peptides, respectively.
These pockets are usually very restrictive to an amino acid residue or a
group of residues that are bound. These residues interact strongly with
these pockets of the MHC-class I molecule and are often referred to as
the “anchor residues” because they confer some degree of specificity to
their associated MHC-I molecule. The rest of the residues in the peptide
interact weakly with the MHC-class I molecule. These interactions are not
with side-chains allowing the side-chains of these residues to be exposed
on the molecular surface for the recognition by the TCR. HLA-B27 has
a strong preference for peptides containing arginine at P2 [20], although
glutamine was also found to reside in P2 in 3% of HLA-B27 ligands
[21]. While the C-terminal residue is the second-most important anchor
residue, it is not as conserved as P2; HLA-B27 ligands may possess a basic,
aliphatic or aromatic residue at this position [22].
Each of the subtypes has a different degree of association to AS
(Table 1). HLA-B*2705, -B*2704, and - B*2707 are linked to AS [23],
while HLA-B*2706 and HLA-B*2709 are the most weakly associated
with the disease [24-27]. These natural variants differ from the B*2705
prototype by one or a few amino acid residues, mostly at positions 114 and
116, and their different degrees of association to AS could be due to the
differential peptide presentation. Differences in the amino-acid residues:
114 and 116, placed at the base of the peptide-binding groove are critical.
The amino acid 116 lies at the bottom of the F pocket, interacting with
the side chain of the peptide C-terminus and the amino acid 114 lies
at the bottom of the pocket D, interacting with residue 3 of the peptide
[28]. B*2709 differs from B*2705 at position 116 by a single amino acid,
aspartic acid (D) to histidine (H) [29]. B*2704 and B*2706 differ by only
two amino acid changes. B*2704 has histidine (H) at 114 and aspartic
acid (D) at 116, while B*2706 has aspartic acid (D) at 114 and tyrosine
(Y) at 116 [30-32]. The AS-associated subtype B*2707 has a tyrosine at
position 116 [33]. Several studies have investigated the features of the
peptide-binding groove and the peptide pools presented by the HLA -B27
subtypes, reasoning that the peptides bound to the associated subtypes
would have different features from the ones, which bind the non-associated
subtypes. In these studies, systematic comparisons of pair-wise associated
and non-associated subtypes (based on populations expressing these
subtypes) were conducted in order to determine which peptide ligands
are presented by the subtypes associated with AS. These studies indicated
that these subtypes differentially associated to AS share about 80% of their
peptide repertoires, and also bind some different ligands showing specific
motifs in the AS-associated subtypes. Therefore, there are some shared
and unique features of the peptides bound by these subtypes which could
correlate peptide presentation and pathogenesis of the disease [34-37].
In summary, non-associated subtypes had a high restriction for nonpolar
C-terminal residues, including aliphatic ones and phenylalanine
as compared to the associated ones, which are able to bind tyrosine.
However, this straightforward correlation has been challenged by the
AS-associated B*2707 subtype, in which, no peptide with tyrosine in the
C-terminal was found [33]. In a more recent and a complete study of the
seven major HLA-B27 subtypes; peptide-bound repertoires and peptides
features, together with molecular stability were examined [38]. The results
showed that peptides derived from AS-associated subtypes had more
diverse C-terminal residues than the non-AS associated subtypes and
this goes with what was shown in previous studies. Also, the residue 116
showed up as a very important feature in defining the peptide binding,
folding and thermodynamic properties of the different subtypes. Subtypes
associated to AS seem to bind better epitopes directly produced in the
cytosol and they were more influenced by the protease ERAP1 (see MHC
class I pathway section).
B*2709 was identified only in 20% of the B27 positive population in
Sardinia and in 3% of the population in continental Italy, but in none of
the AS patients or SpA patients [39]. However, B*2709 was found later
on in some SpA patients, including Tunisians, questioning its ‘nonassociation
with AS [40-42].
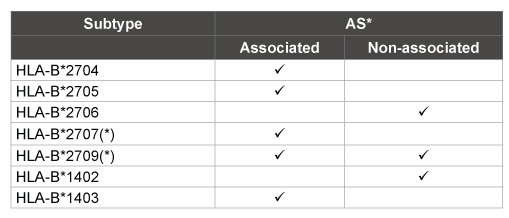
Table 1: Summary of the subtypes and their different association with AS*
(*)Conflict with their association to disease (explained in detail in the
review), AS* (Ankylosing spondylitis)
The weak association between AS and HLA-B*2709, as compared to
HLA-B*2705, was suggested to be in part due to a limited number of
natural ligands bound, exclusively, by this allotype [36]. HLA-B*2709
presents a restriction in the C-terminal residue bound, imposed by its
polymorphism, where mostly all the peptides bound to this subtype
had aliphatic and phenylalanine residues in their C-terminus. All of the
HLA-B*2705-specific peptides possessed arginine, lysine, or tyrosine at
P9. These data support the existence of some specific peptides bound to
HLA-B*2705 which represent a limited set of its bound peptide repertoire
with potential to trigger AS. A study in Sardinia showed that 2 distinct
haplotypes (i.e., blocks of genes in linkage disequilibrium (LD)) that
are transmitted together: A2; B27; Cw2; DR16 which harbors the AS
associated B27 alleles and A32 or A30; B*2709; Cw1; DR12, harboring
the non-associated HLA-B*2709 allele [43]. These findings make feasible
that other genes within the HLA region, besides HLA-B27, may play some
role in conferring susceptibility to AS. Thus, an alternative explanation
for the absence of association between B*2709 and AS in Sardinia, could
be that other alleles of the nearby gene(s) that are in LD with B*2709
confer protection from SpA development [44]. A recent study comparing
peptide repertoires of the 8 most frequent HLA-B27 subtypes has revealed
that quantitative changes in the peptidomes are also important for the
association with AS [45]. The authors used the targeted approach of multiple
reactions monitoring (MRM) mass spectrometry (MS) to precisely look
at low-abundance peptides over the different HLA-B27 allotypes. This
approach allowed the detection of reduced levels of tyrosine as a residue
bound to the C-terminal in B*2709, supporting the previous finding that
this non-associated subtype could bind only two peptides with arginine or
tyrosine in the C-terminal [36]. In part, this finding supports the reason
why B*2709 was previously related to undifferentiated SpA (uSpA) [46],
where the manifestation of the disease does not involve the axial skeleton
but the patients show peripheral manifestations of SpA. These quantitative
differences are important because they could be driving the progression of
the disease and, they may set up the threshold of how much of the antigen
is needed for auto reactive T cells to get selected and activated [45]. It
would be helpful to use this approach to re-evaluate some of the older,
less quantitative data regarding the HLA-B27 subtypes. For example, reexamining
the peptide pool bound to B*2707 would determine whether
there are even a few peptides with tyrosine at the C-terminal.
Therefore, this differential peptide binding may explain the possible
existence of arthritogenic peptides (see below) causing AS. However,
a more refine idea emerged from the elegant x-ray crystallography
structural studies of B*2705 and B*2709 bound to a self-peptide derived
from the vasoactive intestinal peptide, type 1 receptor (pVIPR; sequence
RRKWRRWHL) [47]. pVIPR was displayed in 2 different conformations
when bound to B*2705. In one conformation, the peptide and the heavy
chain of the HLA-B27 molecule are bound by drastically different
interactions, as opposed to the conventional conformation, exclusively
found in the case of B*2709. This led to the speculation that the nonconventional
conformation can alter the potential antigenic surface
presented to the CD8+ T cells and this might generate auto reactivity.
However, the idea of the contribution of dual conformation peptides to
the susceptibility of the disease was challenged by the extended structural
analysis of the B*2704 and the B*2706 subtypes [48]. In this study, the
dual conformation was observed for the non-disease associated B*2706.
In this study, the authors also probed the dynamics of these HLA-B*27
molecules using isotope-edited infrared (IR) spectroscopy, and including
B*2705 and B*2709 as well. Rather than a dual conformation, the results
demonstrated that the disease-associated subtypes B*2704 and B*2705
have a higher conformational flexibility. The heavy chain of the B*2705
complex had already shown an increased conformational flexibility
compared to B*2709 heavy chain, in a previous study [49].
The Arthritogenic Peptide Hypothesis and AS
Based on the canonical function of HLA-B27, the “arthritogenic peptide”
hypothesis suggests that the arthritis in AS patients is a consequence of
HLA-B27 presenting joint-specific peptides to autoreactive CD8+ T cells.
Molecular mimicry, or cross-reactivity between bacterial antigens and
self-peptides, could explain why there may be a break in self-tolerance
after infection with certain pathogens [50,51]. The idea underlying this
mechanism is that self-peptides and bacterial antigens have homology
that makes them cross-reactive, and thus CD8+ T cells would be
primed to the ‘foreign’ antigen and then cross-react against self-peptides
triggering pathogenesis. This hypothesis was strengthened in 1993 when
HLA-B27-restricted cytotoxic CD8+ T cells (CTLs) from the synovial
fluid of AS patients were found to recognize both bacterially infected
and uninfected target cells [52]. This was evidence for the “arthritogenic
peptide” model, where CD8+ T cells restricted to HLA-B27 need to be
isolated from the arthritic joints of patients positive for SpA. Also, high
homology was shown between a self-antigen derived from HLA-B27 itself
and presented by this class I molecule (aa 309-320) and a peptide derived
from Chlamydia trachomatis [53]. Later studies confirmed significant
homology between self-peptides and peptides derived from members of
the Gram-negative Enterobacteriaceae family, including Klebsiella [54],
Yersinia [55] and Salmonella [56-58], that are presented by HLA-B27 [59].
It is unclear whether some of these sequences are generated in vivo or if
HLA-B27 is able to present them directly. The fact that the DNA primase
peptide (211-221) was endogenously processed (from its bacterial
protein) and presented by HLA-B27 shows that this peptide might be the
trigger facilitating the molecular mimicry between Chlamydia and the
homologous HLA-B27 self-ligand (55% homology) and thus, associating
HLA-B27 to disease [56]. The observation that infection with such bacteria
often precedes the onset of AS further supports this hypothesis as well [45].
In summary, these studies show how there could be molecular mimicry
between microbes and self-antigens that could underlie triggering of AS.
Sequence similarities between human self-peptides presented on
HLA-B*2705 and peptides derived from the Hepatitis B virus (HBV)
suggest that molecular mimicry may also play a role in viral infections
[60]. The similarities between HLA-B27 peptides derived from cartilage/
bone proteins and short peptide sequences derived from viruses known to
cause chronic infections [59,21], support this. Since, AS is an inflammatory
autoimmune disease primarily of the joints, it makes sense that the
molecular mimicry between these ‘self ’ and viral proteins could trigger
disease. Another piece of supportive evidence is the extreme prevalence
of the HBV surface antigen (HBsAg) in HLA-B27+ patients with AS,
compared to other SpA patients, HLA-B27_
AS patients and general
population. This may indicate that the high prevalence of this antigen in
AS patients might be associated with the expression of the HLA-B27 gene
and the pathogenesis of the disease through molecular mimicry [61,62].
To reinforce this idea, Sun et al., also assessed the binding affinity between
these viral peptides and HLA-B*2705 by SYFPEITHI epitope prediction
database and Net MHC 3.4 server. This way, the sequences which do
not bind HLA-B*2705 could be distinguished from the HLA-B*2705
candidate epitopes. In this study, it was predicted that among others,
HLA-B*2705 can bind an HBV epitope which has molecular mimicry with
human collagen. Also, crystallography data revealed that HLA-B*2705
can present the viral peptide pLMP2 (RRRWRRLTV), derived from the
latent membrane protein 2 (residues 23-244) of Epstein-Barr virus (EBV)
[63]. This indicates that the concept of molecular mimicry is not limited
between bacterial peptides and self-peptides but also includes viral peptides.
However, the very few shared peptides (3% of the repertoire) by
HLA-B27 and HLA-B14 [64] present a problem in defining the anchor
residues of the arthritogenic peptide(s) [65]. This, along with the evidence
that HLA-B27 disease in transgenic rats [66] does not require CD8+ T
cells, makes it difficult to conclude that AS pathogenesis would be solely
a consequence of cross-reactive CD8+ T cell responses between “self ”
and bacterial or viral mimic peptides. Briefly, another piece of evidence
supporting this conclusion is that the cytokine IL-23 has been recently
shown as a key factor in SpA. The misfolding of HLA-B27 triggers
cellular stress response, followed by the production of IL-23 [67]. CD3+
CD4- CD8- T cells residing at the tendon-bone attachments (entheses)
have been found to respond to IL-23 through their IL-23 receptor, thus
producing the IL-6 IL-7, IL-22 and chemokine (C-X-C motif) ligand 1
(CXCL1), inflammatory mediators. Upon IL-22 production, the signal
transducer and activator of transcription 3 (STAT3) gets activated and
mediates inflammation at the entheses [68].
HLA-B27 and Misfolding
HLA-B27 has a unique peptide binding specificity which favors the
theory that this class I molecule has the ability to present arthritogenic
peptides. However, the lack of evidence supporting the arthritogenic
peptide model in vivo has led to other hypotheses that could explain
HLA-B27 and AS association. HLA-B27 must fold properly in the
endoplasmic reticulum (ER) and associate with B2m and an antigenic
peptide in order for it to be expressed on the cell surface, and therefore
present the antigen to the CD8+ T cells. However, HLA-B27 also has an
aberrant behavior [69]. Compared to other HLA molecules, it exhibits
a slower folding rate and tends to misfold in the ER [70,71], leading
to both stress in the ER and the activation of the unfolded protein
response (UPR) [72]. UPR activates NF-κB and pro-inflammatory
cytokines such as TNFα, IL-6 [73], and IL-23 [74] increase their
expression. The resultant IL-23 can then stimulate a T-helper 17 cell
(Th17) response, which may contribute to the pathogenesis of AS [75].
Because of these unusual HLA-B27 biological properties, the misfolding
hypothesis was proposed [70].
A portion of assembled HLA-B27 heavy chains (HC) were shown to
misfold because of the HLA-B27 B pocket [70] resulting in ER-associated
degradation (ERAD) [76]. When the B pocket of HLA-B27 was replaced
by the B pocket from HLA-A2, B27 HC could fold back [70]. Mear et
al. [70] also compared the peptide-binding and peptide-loading features
of both allotypes. The B27 misfolded HC were degraded in the cytosol,
and overall less HLA-B27 molecules were loaded with peptide. Also, in
the animal models misfolding is exacerbated: in mice due to the absence
of endogenous B2m, and in rats by over expression of misfolded forms
[66]. However, the study of a HLA-B27 transgenic rat model challenged
the misfolding hypothesis [77], showing that an increased B2m expression
could rescue the proper folding of the B27 HC. A reassessment of these
results was done later on, where the HLA-B27 HC up-regulation was
examined [67]. This study showed that extra B2m merely attenuates UPR
activation, but it does not prevent it. Additionally, HLA-B*2707, which
is usually, but not always, associated with AS, [78,79] has similar folding
properties as the non-associated AS subtypes [80]. These properties were
studied in terms of folding efficiency and export rate from the ER to the
cell surface, measured by the acquisition of Endoglycosidase H (EndoH)
resistance. Therefore, given the controversy of the results and the lack of
correlation between the folding properties of AS-associated and nonassociated
subtypes, the evidence suggests that the misfolding hypothesis
is probably not enough by itself to trigger the disease. Therefore, there
must be some other important molecules (e.g., ERAP1 or tapasin,
discussed below) and other HLA-B27 intrinsic properties influencing the
development of AS and/or orchestrating the “right or wrong” behavior of
the HLA-B27, which leads to its association with AS.
Oligomerization and intracellular accumulation patterns have shown a
correlation between biochemical behavior and level of the predisposition
to AS conferred by the different HLA-B27 subtypes [81]. This study
demonstrated that along with an increase in their expression levels,
AS-associated subtypes tend to accumulate in intracellular vesicles and
form more oligomers than the non-associated subtypes. This is the only
study so far showing a complete correlation between subtypes and AS,
although the biological significance is still unknown. The authors argue
that because all subtypes carry cysteine-67 (Cys67) [82] and other Cys
residues important for homodimerization of B27, other factors may
contribute to the association between the formation of oligomers in the
associated subtypes and AS.
HLA-B27 and Cell Surface Homodimers
The canonical form of HLA-B27 at the cell surface is a heterodimer
(HC-B2m) bound to a peptide. HLA-B27 can form polymers and covalent
homodimers in the ER through the cysteine-67 (Cys67) residue in the α1
domain, as well as through other Cys residues [82,71]. Also, homodimers
through just Cys67 can form at the cell surface. These structures are empty
MHC-I molecules and arise by cell surface dissociation of heterodimers
from B2m [83] or – possibly primarily – after dissociation from B2m
after endosomal recycling [84]. The HLA-B27 homodimers hypothesis
arose from this HLA-B27 ability to form homodimers [85], which offers
another explanation for the association of HLA-B27 with AS. Briefly, even
though HLA-B27 homodimers may not acquire appropriate peptides
for cognate interactions with the T-cell receptor (TCR), the killer-cell Ig
-like receptors (KIR3DL2) expressed on natural killer cells and CD4 Th17
cells [86] are able to recognize them. This recognition stimulates IL-17
production, which seems to be a link between the homodimers and the
pathogenesis of AS as it triggers joint inflammation. More importantly,
it has been shown that IL-17 production was increased in the blood
and synovial fluid of patients with SpA, after KIR3DL2 (+) CD4 T cells
expansion and enrichment [75], further supporting the suggested link
between this homodimers formation and AS. However, by itself this
hypothesis does not completely explain the association between HLA-B27
and AS either. All the subtypes have the same cell surface homodimers
formation potential, both their HC dissociated from B2m and free HC
expressed at the cell surface were similar [87]. This is not surprising, since
all the subtypes have a Cys residue at position 67.
Other Allotypes and Non-MHC Factors in AS
HLA-B14
As already stated in the introduction, the strongest association between
a HLA class I molecule and any disease is the association of HLA-B27 with
AS, but there have been other HLA class I molecules linked to AS as well.
HLA-B27 has a strong preference for peptides containing arginine at P2
accommodated in the pocket B [28]. As mentioned above, this pocket B
confers to HLA-B27 unusual unfolding properties that have been linked
to the disease. This cavity is polymorphic among all the class I antigens
and very few allotypes bind arginine at P2 in their peptides, making these
allotypes more interesting in regards to their link to SpAs and AS. One of
these allotypes is HLA-B14 which binds preferentially to peptides with
arginine at P2 [88,64]. Additional evidence in support of the importance
of the arginine at P2 is that the onset of SpAs in gorillas has been correlated
with class I molecules which present peptides with arginine at P2 [89].
The HLA-B*1403 allotype is only found in the populations of
Cameroon and Togo in Africa, where the prevalence of HLA-B27 is rare
and the disease is infrequent, and it was found to be associated with AS
[90,9]. Lopez-Larrea et al. [90] found in the study that in a small cohort
of eight AS patients, four carried B*1403 and one carried B*2705, while
85 healthy controls (used to match for ethnic background) were found
to be B*1402 positive. Given the fact that the size of the cohort used in
this study was small, HLA-B27 is still the statistically strongest MHC-I
associated with AS. B*1402 only differs from B*1403 in position 156: it
is widespread among the Caucasian population and it has never been
found to be associated to AS. These two HLA-B14 molecules, which are
structurally similar but differentially associated to AS (Table 1), have been
investigated as a way of testing the aforementioned hypotheses in a nonHLA-B27
system.
The peptide pool comparisons of the two HLA-B14 subtypes and that
of HLA-B*2705 revealed that the two AS-associated allotypes, B*1403 and
B*2705, share 3% of their peptide repertoires [64]. If the susceptibility to
AS is based on the specific peptide recognition by T cells as is proposed
by the arthritogenic peptide hypothesis [50], it would be expected to
find common peptides with the same structural features between the
two associated subtypes, B*1403 and B*2705. However, both the large
disparity of their peptide repertoires and the lack of binding features
shared by these two allotypes, but not B*1402, argue against (although
do not exclude) a mechanism of spondyloarthritis by specific ligands of
B*2705 and B*1403. The joint finding of a few shared ligands and crossreactive
CTL clones between HLA-B27 and HLA-B14 [64] suggests that
B*1403 and B*2705 present either some shared peptides with the same
antigenic features or distinct peptides showing antigenic mimicry.
A study comparing stability, maturation, assembly and folding
properties of HLA-B*1402 and B*1403 to those of B*2705 [91], revealed
that B*1402 and B*1403 have similar folding rates, faster and more efficient
than B*2705. However, some unfolded HC from both B14 subtypes
remained in the ER with a longer half-life than B*2705, indicating that
their export rates are slower than B*2705. The finding of some Endo-H
resistant HC for both B14 subtypes indicates that the heterodimers
partially dissociate after exiting the ER. Thermostability and interaction
with tapasin (a chaperone which brings peptides to MHC-I molecules)
was highest for B*2705 and lowest for B*1403. Altogether, this suggests
that the B*1402-bound peptides and especially the B*1403-bound
peptides were less optimized than those of B*2705. Because the biological
features of B*1403 differ more from B*2705 than from B*1402, it does not
seem that obvious that the underlying association with AS could be driven
by the same biological properties in different associated class I allotypes.
Therefore, it is worth, reassessing the significance of B*1403 and B*2705
sharing a low level of peptides and T cell epitopes. An obvious alternative,
which does not oppose to the comparative biology of the three allotypes,
might be a shared ligand of these two associated allotypes. However, in
the absence of a formal demonstration of this shared arthritogenic peptide
and the incomplete explanation of the association with AS, by the other
hypotheses, other non-MHC factors have to be evaluated.
Other Components of the MHC-I Pathway
MHC class I pathway
Endogenous proteins are primarily degraded by the proteasome [92]
which generates mature MHC-I epitopes usually between 8-11 amino
acids long, depending on the class I molecule. In the case of HLA-B27,
crystallography studies revealed nonamers as the most common bound
peptides [93]. Approximately 10-15% of peptides are too long to bind
directly to MHC-class I [94-96] and must undergo subsequent N-terminal
trimming in the cytosol and/or ER. The peptides are translocated from
the cytosol to the ER through the transporter associated with antigen
processing (TAP) [97]. The endoplasmic reticulum aminopeptidase 1
(ERAP1) is the main responsible protease of N-terminal trimming of
antigenic precursor peptides in the ER [98-100]. Endoplasmic reticulum
aminopeptidase 2 (ERAP2) is also capable of this N-terminal trimming
[101]. Following its proper folding, MHC-class I binds to B2m and is
incorporated into the peptide-loading complex (PLC) [102]. The PLC,
which consists of TAP, tapasin, calreticulin, calnexin, and ERp57, helps
load MHC class I molecules with their peptide cargo [103].
ERAP1
Although HLA-B27 remains a dominant risk factor in susceptibility
to AS, non-MHC molecules and other factors have been linked to the
susceptibility of the disease [104]. In the last few years, these other factors
are attracting more attention and more research has been conducted to
better understand how the susceptibility to SpAs -and in particular to
AS- is driven. ERAP1 was estimated to be the strongest non-MHC gene
associated with AS, contributing to the association to the disease with a
risk of 26% [105].
Different AS-associated ERAP1 single nucleotide polymorphism
(SNPs) have been reported [106,107]. These different natural variants
of ERAP1 have revealed different peptide length preferences as well as
changes in the enzymatic activity [108] and stability of HLA-B*2704-
peptide complexes [109]. García-Medel and colleagues showed that
there is a correlation between ERAP1 polymorphisms associated with
AS susceptibility, a efficient peptide trimming by this protease and high
stability of HLA-B27, whereas protective polymorphisms against AS
were associated to an attenuated activity of ERAP1, less active trimming,
and decreased molecular stability of the class I molecule, suggesting less
optimized HLA -B27 peptidomes. These findings suggest that the way
in which ERAP1 and HLA-B27 interact is important in AS, and were
consistent with those from a previous study where the SNP rs30187
(K528R) is a protective variant associated with reduced enzyme activity
in vitro [110]. The SNP K528R, which is away from the enzyme’s active
site, controls the enzyme open-closed conformations, leading to more
closed conformers which are consistent with decreased enzymatic activity
[108]. On the contrary, the natural ERAP1 polymorphism predisposing
to AS: R528K altered the expression levels of many HLA-B*2705-bound
peptides accounting for the association of this SNP with AS [111].
Currently, the main role of ERAP1 in MHC-I-associated AS seems to be
through its effects on the MHC-bound peptidome [112]. However, due
to ERAP1 involvement in angiogenesis [113] and macrophage activation
[114], the existence of other inflammatory and immune pathways linked
to AS through and indirect effect or ERAP1 cannot be ruled out.
Dendritic cells isolated from HLA-B27 AS patients expressed more
ERAP1 than those from healthy individuals [115]. This finding was
of interest since the SNPs identified in patients with AS by Harvey et
al. [107] localized upstream the gene in a regulatory region, possibly
impacting ERAP1 expression levels. Similarly, in a more recent study,
the antigen presenting cells had their levels of gene expression affected
by the SpA-associated ERAP1 polymorphisms. In dendritic cells and
lymphoblastoid B cells isolated derived from these SpA patients, there
was an association between ERAP1 SNPs predisposing to disease
and higher ERAP1 mRNA expression levels, as well as higher ERAP1
transcripts or protein levels [116].
All these data point out that there is a correlation between: ERAP1
SNPs and predisposition to AS and higher ERAP1 expression both at
mRNA levels and protein levels, as well as a more active enzyme. Since this
protease plays an important role in the antigen processing and presentation
in the MHC-I pathway, it is a critical link between susceptibility to AS
and generation of peptide antigens to be presented by the MHC class I
molecule HLA-B27.
Tapasin
Tapasin is a chaperone which binds HLA class I molecules [117],
brings other members of the PLC onto TAP [118], shapes the HLA class
I repertoire [119,220], increases the stability of HLA class I molecules
[121-123] and influences both quantitatively and qualitatively the peptide
repertoire [124].
The interactions between tapasin and HLA-B27 are mediated by the
amino acids at positions 114, 116, and 152 in the peptide-binding groove
of HLA-B27. These positions are key for these two molecules to interact
with each other [125]. Interestingly, the B27 subtypes associated and nonassociated
with AS, differ at some of these positions. B*2705 and B*2709
are only different at the amino acid position 116, D116H [29]. This amino
acid is located at the bottom of the F pocket, binding the C-terminus of
the peptide [126]. B*2704 and B*2706 are different at positions 114 and
116: H114D and D116Y [30-32]. B*2707, an associated subtype, lacks D
in position 116 (where a Y lies instead) like B*2706 and B*2709 which are
not associated with AS. B*1402 and B*1403 differ only in position 156,
where B*1402 has leucine and B*1403 arginine. This position has been
suggested to affect the interaction between TAP and MHC-I [127], which
is mediated by tapasin [117]. Experimental and theoretical research has
proposed that the F pocket is the binding region of tapasin [128-131].
Since this pocket accommodates the C-terminal residue of the peptide
bound to class I, one of the anchor residues, this suggests that changes
in the interaction between tapasin and MHC-I could somehow drive the
susceptibility to AS.
Some studies (discussed in the following paragraphs) have looked
at how polymorphism changes in the B27 and B14 subtypes (already
reviewed in the HLA-B14 section) may influence their interactions with
tapasin, their dependency on tapasin to present peptides and what would
be the consequences for AS susceptibility.
The tapasin dependence of a particular class I allotype was predicted
using combinations of in silico and experimental approaches. These
approaches used the sequence and crystal structure of a particular class
I molecule. These approaches have demonstrated that B*2705 is more
dependent on the chaperone than the conformationally stable B*2709
[132], in order to remain structured or properly folded and to bind peptides.
A more unstable class I molecule would be more prone to misfolding
and aggregation, thus being more susceptible to trigger pathogenesis.
However, there is some controversy as to whether all the associatedsubtypes
have greater tapasin dependence than the non-associated
subtypes. Some studies have analyzed B*2705-peptides complexes at the
cell surface of tapasin-deficient cells and found that expression of B*2705
is independent of tapasin [124,121,133]. Compared to B*2705, B*2704
(another AS-associated subtype) is relatively dependent of tapasin for
its surface expression [134]. However, in terms of maturation, these two
associated molecules showed a similar tapasin dependency in this study.
Both subtypes showed an inherent tendency to misfold, when tapasin is
not present and too accumulated in the ER with relatively slow export
to the cell surface. In contrast, B*2706 showed no accumulation in the
ER and faster folding in the absence of tapasin. These results link tapasin
to the misfolding hypothesis discussed above, as a potential explanation
for the susceptibility to AS. It is worth noting though that B*2709, not
associated to AS, matures similarly to B*2704 and B*2705, at least in the
presence of tapasin [125]. In a different study, B*2709 was found to mature
differently from B*2704 and B*2705 [80]. These two studies contradict
each other and part of the reason for the different results could be that
different cell lines were used to carry out these experiments. Again, the
controversy and an imperfect correlation between the non-associated
and associated subtypes with tapasin, do not explain the totality of the
predisposition to the disease.
Microbiota and AS
The human microbiota, which represents the totality of microorganisms
residing in the human body, has been recently presented as another factor
in the etiopathogenesis of SpA. HLA-B27 and altered cecal microbiota
have been associated [135]. The number of bacterial cells is 10-fold
greater than human cells, being up to 100 trillion cells in the gut [136].
These organisms have been implicated in different aspects of the gut:
maintaining homeostasis in a healthy state [137], regulating energy supply,
controlling colonic pH, preventing the invasion of pathogens, and keeping
intestinal health [138,139]. Bacterial dysbiosis promotes inflammation
and may confer the development of human disease, linking bacterial
composition and the immune system [140]. The intestinal microbiome
in healthy individuals is now available thanks to the 16S ribosomal RNA
(16S rRNA) sequencing technology. Nine divisions of bacteria comprise
the microbiome and the majority of the species belong to four of them:
Bacteriodetes, Firmicutes, Proteobacteria, and Actinobacteria [141].
Distinct clusters or “enterotypes” of bacteria that differ in their composition
and function can also compose the human gut microbiome. The genus
Bacteriodetes dominates the enterotype 1; Prevotella, the enterotype 2
and, the enterotype 3 is dominated by Ruminococcus [142]. Species like
Prevotella have been found to be increased in HLA-B27 transgenic rats
and some other are decreased compared to wild-type rats [143].
The role of endogenous flora in the pathogenesis of AS has gained
more relevance over the years, and increasing evidence supports the
idea that there is a link between bacterial dysbiosis, HLA-B27, and AS.
Inflammatory bowel disease (IBD) and AS have considerable clinical
overlap and there is also an understanding that bowel flora play a role
in IBD [144]. Around 7% of patients with AS have IBD, and 50-60% of
AS patients have subclinical gut inflammation [145]. There are also some
reports relating SpA and bacterial flora. Chlamydia for example triggers
ReA, within the group of SpA, by inducing the expression of interleukin
23 (IL-23) in infected cells [146]. Several studies have reported distinct
microbial colonization between AS patients and healthy controls,
reviewed elsewhere [144]. Also, HLA-B27 has been proposed to alter
the gut microbiome and to be linked to the development and severity of
ReA. These patients are inefficient at eliminating the causative bacteria
[147]. Human monocyte cells where HLA-B27 was expressed, showed
more impairment to handle intracellular replication of Salmonella,
suggesting that the shape of the intestinal microbiome may be influenced
by intracellular effects of HLA-B27 [148]. Some class I heterodimers tend
to dissociate, presumably in the endosomal compartment after exiting the
ER. This class I heterodimers that are in the endosomes are likely ones that
have left the cell surface. This was showed for B*1403 and B*1402 where
the percentage of Endo-H resistant free HCs increased within the time,
along with a decrease in the percentage of HLA-B14 heterodimers [91].
This was interpreted as B14 having a less optimized repertoire of bound
peptides, resulting in lower peptide stability. Indeed, B*1403 showed less
binding of HC to tapasin over the time and less thermostability for its
MHC/peptide complexes than B*1402. Most likely, following endosomal
recycling, the B14-peptide complexes dissociate at late maturation
stages as it has been described for B27 [84] and as a consequence of a
less optimized repertoire of bound peptides. This dissociation could be
triggered by the endosomal acidic pH, allowing peptide exchange in this
compartment [149], where either endogenous or pathogenic peptides can
be bound to class I molecules to be presented to CTLs. B*2705 did not
show an increase in endo-H resistant free HCs [91], but their existence
in HLA-B27+ AS patients cannot be ruled out. Thus, it could also be
plausible that HLA-B27 is recycling to the phagolysosomes (a more acidic
endosomal compartment) -where Salmonella resides- and because of its
improper folding is unable to bind Salmonella peptides properly to present
to CD8+ T cells, leading to a lack of immune response against this pathogen
and therefore facilitating a persistent infection. The impaired elimination
of microbes triggering ReA by HLA-B27+ monocytes may explain the
persistence of these microbes in HLA-B27+ individuals susceptible to
ReA [150,151]. Class I molecules can present exogenous antigens in a
process called cross-presentation [152] and studies in mice have shown
that dendritic cells, which are antigen professional presenting cells, can
process Salmonella antigens and elicit Salmonella-specific CD8+ T cells
responses [153]. In the case of HLA-B27+ AS patients, I postulate that
this cross-presentation pathway could be defective and further research
could facilitate the understanding of how the microbiota, HLA-B27 and
probably other class I antigens, and AS are related.
In their hypothesis, Rosenbaum and Davey proposed that HLA-B27
shapes the human endogenous flora which causes AS [154]. However, a
more recent study argues that immune dysfunction drives dysbiosis since
immunological changes occur in the gut prior to any detectable microbial
changes [155]. This does not rule out the possibility that HLA-B27 shapes
the microbiome, but rather that immune dysfunction underlies these
changes.
Penttinen et al. [148] also showed that glutamic acid at position 45 in the
B pocket drives this reduced capacity to handle intracellular replication of
Salmonella. This B pocket influences the folding properties of HLA-B27,
which can lead to UPR, as already mentioned. However, Penttinen et al.
[148] did not find evidence for an ongoing UPR. Along these lines, in a
more recent study, data suggested that there is HLA-B27 misfolding in
the gut of HLA-B27+ AS patients, together with autophagy activation
rather than a UPR [156]. Autophagy and intestinal modulation of IL-
23 in AS, appear to be associated. Also, AS patients with subclinical gut
inflammation presented a local excessive production of IL-23 [157].
Autophagy is a process which helps in the maintenance of cellular
homeostasis by degrading cellular constituents [158]. It is involved
in host cellular defense against pathogens [159] and eliminates
improperly folded proteins [160]. This targeting of improperly folded
proteins for degradation occurs in the ER, similar to the UPR process
[161] and it was suggested, that the inability to demonstrate UPR in
all mentioned above studies could be due to compensation by excess
autophagy [162].
HLA-B27 and TCR repertoire
Another theoretical mechanism for B27 association with AS is an
altered TCR repertoire due to different positive or negative selection (and/
or the development of regulatory T cells –Tregs-) on B27 in the thymus.
Briefly, during positive selection in the thymus, only the thymocytes
that interact appropriately (not too strongly or too weakly) with MHC-I
molecules (also MHC-II) will receive a ‘survival signal’, thus the selected
T-cells will have affinity to interact with MHC peptide complexes and to
effect immune responses. Negative selection removes thymocytes capable
of binding in a very strong way with ‘self ” MHC peptides, thereby selftolerance
can be maintained in order to avoid autoreactivity. It could
also be that autoreactive T cells are redirected into Tregs cells [163]. One
could speculate that if either one of these processes fail, it could lead to
autoimmunity. If negative selection fails, then autoreactive T cells will
not be eliminated, thus creating autoimmune disease. If in this case
the selection is done on B27, autoreactive T cells would recognize selfpeptides
presented by the MHC-class I molecule, leading to AS. In the
case of positive selection failure, T cells would not recognize foreign
MHC-peptide complexes. The depletion of T cells would lead to a
situation where a bacterial or viral infection would become persistent,
triggering accumulation of complexes and MHC molecules. This
accumulation of complexes leads to inflammation and AS, as discussed
above (see HLA-B27 and misfolding section), even in the absence of T
cells. Also if Tregs cells are not generated normally, then this could lead to
autoimmunity. The contribution of altered TCR repertoires in the context
of ReA and SpA was recently reviewed [164].
Conclusion
HLA-B27 is a peculiar MHC class I molecule with features that make
it suitable to be linked with an autoimmune disease such as AS. It is a
HLA class I molecule that binds and presents immunodominant peptides
to cytotoxic T cells during important infections, such as; influenza, HIV,
EBV, and hepatitis C [45]. As with all MHC molecules, B27 presents
peptides but it also sets the perfect environment for T cell cross-reactivity,
due to the high homology between self–peptides derived from the
HLA-B27 molecule itself and microbial peptides. It tends to misfold in
the ER producing stress through UPR, and the accumulation of free heavy
chain allows for the formation of homodimers, at the cell surface. These
features have been the main focus of the different hypotheses proposed
to explain the link between HLA and AS. However, as evidenced by the
comparative studies between different allotypes associated with AS (as
e.g., HLA-B*2705 and HLA-B*1403) and between the different HLA-B27
subtypes; none of these hypotheses completely explain the association of
these HLA-class I molecules with AS. The underlying mechanism of the
association with AS seems more of a combination of the effects of many
HLA, non-HLA genes (mainly covered in this review) and other factors,
which within the last few years have gained more and more attention due
to their shown link with AS. This together is taking HLA-B27 away from
being the only risk factor to the susceptibility to AS (Figure 2).
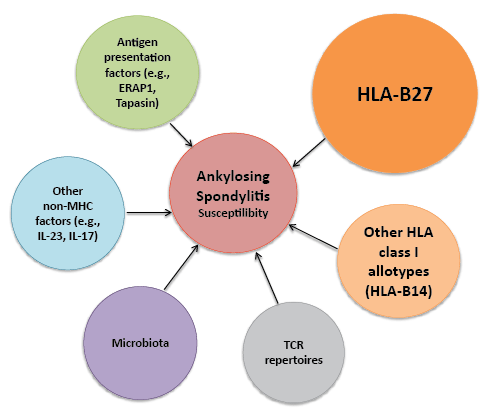
Figure 1: Contributions to AS susceptibility: Contributors to AS susceptibility include HLA-genes, non-HLA genes, antigen presentation factors,
other non-MHC factors (not involved in the antigen presentation MHC-I pathway), the microbiota and the TCR repertoires. All these factors have taken
HLA-B27 away from being the only risk factor to the susceptibility to AS. Thus, AS is a multi-factorial disease influenced by multiple molecules.
Dissecting each of these hypotheses helps us to better understand
the mechanisms underlying AS pathogenesis. However, because of the
multiple molecules and mechanisms influencing the susceptibility to
AS and the fact that all are involved in immune responses, it is worth
considering that multiple of these mechanisms influence whether or not
AS develops. Further study of these players would help us to elucidate the
mystery behind the association of these components and HLA-B27 with
AS, a disease that has been researched for the last 40 years.
Acknowledgment
I thank Dr. Kenneth L. Rock (UMass Medical School, Worcester,
MA), Dr. José Antonio López de Castro (Centro de Biología Molecular
Severo Ochoa, Madrid, Spain), Dr. Robert A. Colbert (National Institute
of Health, Bethesda, MD), Dr. Kenneth Chrobak (Pfizer - Rinat Cell
Engineering Facility, South San Francisco, CA) and Barry Kriegsman
(MD, PhD student, UMass Medical School, Worcester, MA) for revising
the manuscript and providing critical editing and intellectual content.
Conflicts of Interest
None